Миелодепрессия что это такое
Миелодиспластический синдром — Википедия
Миелодиспластический синдром (МДС) — группа гетерогенных клональных заболеваний, характеризующаяся наличием цитопении в периферической крови, дисплазии в костном мозге и риском трансформации в острый лейкоз.
МДС сегодня является одной из самых сложных проблем гематологии. Лишь недавно лечение МДС вышло за рамки поддерживающей терапии, проводившейся с целью облегчения симптомов.
МДС — это патология старшей возрастной группы. 80 % случаев МДС приходится на лица старше 60 лет. МДС в детском возрасте встречается крайне редко. В европейских странах среди лиц 50-69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше — 150 новых случаев на 1 млн населения. Заболеваемость МДС в РФ в среднем составляет 3-4 случая на 100 тыс. населения в год и увеличивается с возрастом. [2]
Первичный (идиопатический) тип — 80-90 % случаев, вторичный (вследствие предшествующей химиотерапии и др. факторов) — 10-20 %. Большинство (80 %) случаев МДС являются первичными — идиопатическими или de novo (лат. — вновь появившийся, новый).
Вторичный МДС является значительно более неблагоприятным и резистентным к лечению типом МДС, обладающим заведомо более худшим прогнозом в сравнении с первичным МДС. 10-20 % случаев МДС возникают вследствие предшествующей химиотерапии по поводу других новообразований. К препаратам, обладающим доказанной способностью повреждать геном с последующим развитием МДС, относятся алкилирующие агенты (циклофосфан), ингибиторы топоизомеразы — противоопухолевые агенты растительного происхождения (топотекан, иринотекан и др.), антрациклины (доксорубицин) и подофиллотоксины (этопозид). К МДС также могут приводить радиотерапия и контакт с токсическими материалами.
- Факторы риска, первичный МДС
- Факторы риска, вторичный МДС
Предшествующая химиотерапия онкологического заболевания или после ТКМ.
Прогноз: 5-летняя выживаемость при МДС не превышает 60 %. Трансформация в острый лейкоз ~30 % случаев.[3][4]
Причины МДС до конца не известны. В основе патогенеза МДС лежит воздействие повреждающих факторов на полипотентную стволовую клетку, приводящее к появлению в ней генетических аномалий, а также феномена гиперметилирования ДНК.
Указанные нарушения приводят к нарушению продукции клеток миелоидного ростка и появлению миелобластов в костном мозге и периферической крови, вследствие чего появляются диспластические изменения в зрелых клетках и их функциональная недостаточность, приводящие к описанным клиническим проявлениям.
Феномен гиперклеточности костного мозга на фоне периферической цитопении объясняется ускоренным апоптозом аномально пролиферирующих клеток костного мозга.[5]
МДС отличает отсутствие типичной клинической картины. Симптоматику МДС составляют последствия дисмиелопоэза, то есть цитопении: анемия, нейтропения и тромбоцитопения (анемия Hb меньше 110 г/л, нейтрофилы меньше 1,800 на 1 микролитр крови; гематокрит меньше 36 % эритроцитов в общем объёме крови в организме; тромбоциты меньше 100,000 на 1 микролитр крови).
Наиболее часто МДС манифестирует цитопениями, главным образом анемией. При этом необходимо дифференцировать МДС от железо- или B12- дефицитной анемии, постгеморрагической анемии, анемии при хронических заболеваниях и онкологии или связанной с хронической почечной недостаточностью, а также апластической анемией, пароксизмальной ночной гемоглобинурией. У 10 % пациентов имеются признаки инфекции, а у несколько меньшей доли пациентов болезнь проявляется кровотечениями.
В связи с этим диагностика МДС базируется исключительно на лабораторно-инструментальных методах, из которых ключевыми являются полный клинический анализ периферической крови, некоторые биохимические исследования и морфологический анализ аспиратов и биоптатов костного мозга.
Дифференциальная диагностика МДС также затруднена в силу множества состояний, имеющих общие с МДС клинико-лабораторные проявления.
Для анализа изменений в периферической крови проводится полный, с подсчетом ретикулоцитов (ускоренный эритропоэз с макроцитозом в ответ на гемолиз и острую кровопотерю приводит к увеличению ретикулоцитов), тромбоцитов и лейкоцитов клинический анализ крови. Типичными находками являются изменения формы клеток, патологические включения и уменьшение числа клеток одного или нескольких ростков кроветворения.
Другим ключевым с точки зрения диагностики МДС оценки прогноза и выработки тактики лечения больных исследованием является морфологическое, иммуногистохимическое и цитогенетическое исследования ткани костного мозга. Исследование костномозгового пунктата в этом отношении является несравненно более информативным, чем определение морфологического состава периферической крови.
Используются два способа получения материала: 1) аспирационная биопсия костного мозга и 2) трепанобиопсия из гребня подвздошной кости.
При цитологическом исследовании костного мозга (миелограмма) можно оценить наличие дисплазии миелоидного ростка.
Дизэритропоэз: многоядерность Костный мозг больного рефрактерной сидеробластной анемией: кольцевые сидеробласты- Дизэритропоэз
Ядро
- Межъядерные цитоплазматические мостики
- Кариорексис
- Многоядерность
- Баббл-формы
- Мегалобластоидность
Цитоплазма
- Кольцевидные сидеробласты
- Вакуолизация
- Положительная реакция с Шифф-йодной кислотой
- Дисгранулопоэз
- Маленькие или чрезмерно большие клетки
- Гиполобулярность ядер (псевдо-Пельгер-Хюит аномалия)
- Неравномерная гиперсегментация
- Гипо-/агрануляция
- Гранулы псевдо Чедиак-Хагаси
- Палочки Ауэра
- Дисмегакариопоэз
- Микромегакариоциты
- Гиполобулярные ядра
- Многоядерность
Гистологическое исследование костного мозга (трепанобиопсия) позволяет оценить архитектонику костного мозга, диффузный или очаговый характер изменений в нём, изучить соотношение кроветворной и жировой ткани, выявить атипичные клетки и т. п. Аспирация костного мозга при стернальной пункции так или иначе нарушает структуру костного мозга и не исключает примешивание к пунктату периферической крови. В связи с этим выполнение трепанобиопсии обязательно для подтверждения диагноза МДС.
Биохимические исследования обмена железа, содержания витамина В12 и фолиевой кислоты, иммунологические пробы призваны помочь провести дифференциальную диагностику с анемиями иного генеза, с учётом того, что у 80 % пациентов с МДС отмечается анемия.
МДС следует дифференцировать с другими онкогематологическими заболеваниями, включая острые и хронические лейкозы, а также лимфопролиферативные заболевания.
Часть изменений, свойственных МДС (в частности, моноцитоз, цитопенические нарушения), могут отмечаться при некоторых инфекционных процессах.
При отравлении тяжелыми металлами могут отмечаться изменения эритроцитарного ростка, сходные с таковыми при сидеробластных анемиях.
У пациентов с наследственными цитопениями рекомендуется проведение дополнительного генетического исследования, которое поможет выявить анемию Фанкони и врождённый дискератоз.
Повторяющиеся хромосомные аномалии при MDSПри диагностике хромосомные аномалии обнаруживаются у 40-70 % пациентов с первичным MDS и у 95 % пациентов, MDS которых связан с терапией (вторичный).
К наиболее часто встречающимся при MDS цитогенетическим аномалиям относятся del(5q), −7 и +8.[6]
| Группа риска | Кариотип (22 группы) | Средняя выживаемость (мес) | Время, к которому 25 % пациентов развили ОМЛ |
|---|---|---|---|
| Благоприятная | 5q-, 12p-, 20q-, +21, -Y, 11q-, t(11(q23)), норма; любые 2 аномалии, включающие 5q- | 51 | 71,9 |
| Промежуточная-1 | +1q, аномалии 3q21/q26, +8, t(7q), +19, −21, любая др. одиночная поломка; любые двойные аномалии, не затрагивающие хр. 5q и 7 | 29 | 16 |
| Промежуточная-2 | -X, −7 или 7q-, любые двойные аномалии с −7 or 7q-, комплекс из 3х аномалий | 15,6 | 6 |
| Неблагоприятная | Более 3х аномалий | 5,9 | 2,8 |
Минимальные диагностические критерии[править | править код]
Минимальные диагностические критерии МДС включают обязательные диагностические условия (цитир. По NCCN, 2009)[7] — стабильная цитопения не менее 6 месяцев, (за исключением случаев, когда цитопения сопровождается специфическим кариотипом или дисплазией двух ростков кроветворения — в этих случаях длительность стабильной цитопении должна составлять не менее 2 месяцев).
- исключение других заболеваний, которые могут стать причиной развития дисплазии или/и цитопении.
В дополнение к этим двум диагностическим условиям для установления диагноза МДС необходимо соответствие хотя бы одному из трёх основных критериев:
- дисплазия (≥ 10 % клеток одного или более из трёх основных ростков кроветворения в костном мозге).
- содержание бластов в костном мозге 5-19 %.
- специфический кариотип, например делеция (5q), делеция (20q), +8 или −7/делеция (7q).
Кроме того, для диагностики МДС используются дополнительные критерии, в том числе результаты проточной цитометрии, гистологического и иммуногистохимического исследования костного мозга, выявления молекулярных маркеров.
Морфологическое исследование биоптатов, полученных путём билатеральной трепанобиопсии, является полезным, помимо верификации диагноза самого МДС, с точки зрения дифференциальной диагностики с лимфопролиферативными и другими миелопролиферативными заболеваниями.[7]
Дифференциальная диагностика проводится с:
- А. Мегалобластными анемиями (заболевания, характеризующиеся изменениями морфологии клеток к.м. вследствие нарушения синтеза ДНК. Более 90 % — В-12 и фолиево-дефицитные анемии).
После начала терапии витамином В-12 или фолиевой кислотой в анализе крови выявляется ретикулярный криз на 5-7 сутки и повышение показателей красной крови, что нехарактерно для больных рефрактерной анемией. Изменения кариотипа клеток костного мозга не встречаются при мегалобластных анемиях.
Апластическая анемия может быть врождённой, приобретённой и идиопатической. Врождённая апластическая анемия — анемия Фанкони сочетается с другими генетическими аномалиями (кожная пигментация, гипоплазия почек, микроцефалия), приобретённая связана с действием химических и физических агентов, инфекциями, нарушениями обмена веществ.
Для АА нехарактерны изменение кариотипа, гиперклеточный костный мозг.
- В. Анемии при ХПН.
- В. Анемии при хроническом активном гепатите характерно выявление маркеров вирусных инфекций, гепатоспленомегалия, клиническая картина хр. гепатита, изменения биохимических показателей крови (метаболизма билирубина, функции печени).
Разработка этой системы классификации франко-американо-британской группой была начата в 1976 году и позже, в 1982 году, она приняла свой окончательный вид.
В основе классификации лежит ключевой для МДС синдром — рефрактерная, то есть устойчивая к лечению препаратами витамина В12 и фолиевой кислоты, анемия (РА). Четыре типа РА являются последовательными стадиями, с нарастанием тяжести МДС, что имеет своё отражение в прогнозе выживаемости. В этой связи появление в КМ бластов резко меняет прогноз выживаемости в худшую сторону.
| Тип МДС | Бластов в периферической крови | Бластов в КМ | Другие патологические изменения | Выживаемость (лет) |
|---|---|---|---|---|
| Рефрактерная анемия (РА) | меньше 1 % | меньше 15 % кольцевых сидеробластов | меньше 5 % | 4,2 |
| РА с кольцевыми сидеробластами | меньше 1 % | больше 15 % кольцевых сидеробластов | меньше 5 % | 6,9 |
| РА с избытком бластов (РАИБ) | меньше 5 % | 5-20 % | — | 1,5 |
| РАИБ в стадии трансформации | больше 5 % | 21-29 % | Возможно наличие палочек Ауэра в КМ | 0,6 |
| ХММЛ | меньше 5 % | меньше 20 % | Моноциты больше 1х109/л | 2,4 |
Французско-американско-британская классификация позволяет отнести пациента к той или иной группе миелодиспластических синдромов в зависимости от морфологических показателей. Группа миелодиспластических синдромов включает пять заболеваний: рефрактерную анемию, рефрактерную анемию с кольцевыми сидеробластами, рефрактерную анемию с избыточным количеством бластов, рефрактерную анемию с избыточным количеством бластов на стадии трансформации и хронический миеломоноцитарный лейкоз. Согласно французско-американско-британской номенклатуре пациентам, у которых содержание бластов в костном мозге превышает 30 %, устанавливается диагноз острого миелоидного лейкоза.
В данной классификации хронический миеломоноцитарный лейкоз относится к группе миелодиспластических синдромов, несмотря на то, что это заболевание часто характеризуется признаками миелопролиферативного расстройства.[8]
В 2002 году Всемирная организация здравоохранения предложила новую классификацию миелодиспластических синдромов[9][10][11] в 2008 году были сделаны предложения по её пересмотру.[12][13]
Подгруппы, выделяемые в классификации ВОЗ включают: рефрактерную анемию и рефрактерную анемию с кольцевыми сидеробластами, рефрактерную цитопению с множественной дисплазией, рефрактерную анемию с избыточным количеством бластов-1 (содержание бластов в костном мозге составляет менее 10 %), рефрактерную анемию с избыточным количеством бластов-2 (содержание бластов в костном мозге превышает 10 %), синдром делеции 5q и миелодиспластический синдром неклассифицированный (с наличием или отсутствием кольцевых сидеробластов).
Пациенты, ранее классифицировавшиеся как страдающие хроническим миеломоноцитарным лейкозом, относятся к группе миелодиспластических синдромов/миелопролиферативных заболеваний.
Синдром делеции 5q, выделяемый в классификации Всемирной организации здравоохранения в отдельную подгруппу, характеризуется изолированной делецией 5q[14][15][16] и содержанием бластов в костном мозге меньше 5 %, часто в сочетании с тромбоцитозом.
| Тип МДС | Изменения в крови | Изменения в КМ |
|---|---|---|
| Рефрактерная анемия (РА) | Анемия, меньше 1 % бластов | Дисплазия эритроидного ростка, меньше 5 % бластов |
| Рефрактерная анемия с кольцевыми сидеробластами (РАКС) | То же, что и РА | то же, что и РА, ≥ 15 % кольцевых сидеробластов |
| Рефрактерная цитопения с многоростковой дисплазией (РЦМД) | Цитопения по 2-3 росткам, меньше 1 % бластов | Дисплазия в больше 10 % клеток 2 или 3 ростков, меньше 5 % бластов, меньше 15 % кольцевых сидеробластов |
| Рефрактерная цитопения с мультилинейной дисплазией и кольцевыми сидеробластами (РЦМД-КС) | То же, что и РЦМД | То же, что и РЦМД, ≥ 15 % кольцевых сидеробластов |
| Рефрактерная анемия с избытком бластов, тип I (РАИБ-1) | Цитопении, меньше 5 % бластов | 5-9 % бластов |
| Рефрактерная анемия с избытком бластов, тип II (РАИБ-2) | Цитопении, 5-19 % бластов | 10-19 % бластов |
| Синдром 5q- | Анемия, нормальное или повышенное содержание тромбоцитов | Нормальное или увеличенное количество мегакариоцитов с гипосегментированными ядрами; изолированная делеция 5q31 |
| МДС неклассифицированный (МДС-Н) | Цитопения | Унилинейная дисплазия в нейтрофильном или мегакариоцитарном ростках, Бласты менее 5 %, Палочки Ауэра отсутствуют |
Всемирная организация здравоохранения предложила исключить рефрактерную анемию с избыточным количеством бластов на стадии трансформации из группы миелодиспластических синдромов (диагноз острого миелоидного лейкоза устанавливается, если содержание бластов в костном мозге превышает 20 %, тогда как ранее для установления этого диагноза содержание бластов должно было превышать 30 %). Однако миелодиспластические синдромы отличаются от вновь диагностированного острого миелоидного лейкоза не только содержанием бластов, но и течением заболевания, обусловленным определёнными биологическими свойствами. Кроме того, эти группы заболеваний обычно отличаются и по частоте терапевтических ответов.
Шкала IPSS (International Scoring Prognostic System — Международная шкала оценки прогноза) была разработана в 1997 году с целью дать специалистам, помимо классификации, практический инструмент по оценке прогноза и выбора тактики лечения для пациентов с впервые установленным диагнозом МДС (то есть не подходит для прогноза уже леченных пациентов с МДС).
Вторичный МДС оценивается как изначально неблагоприятный, автоматически попадающий в категорию наиболее высокого риска согласно IPSS.
Тремя факторами, которые учитывает IPSS для оценки прогноза, являются количество бластов, категория цитогенетического риска и количество поражённых цитопенией линий.
Трактовка результатов суммирования баллов по этим трем параметрам представлена в таблице:[16]
| Количество балов | ||||||
|---|---|---|---|---|---|---|
| Прогностический фактор | 0 | 0,5 | 1,0 | 1,5 | 2,0 | |
| Бласты в костном мозге | меньше 5 % | 5-10 % | — | 11-20 % | 21-30 % | |
| Прогноз с учётом характеристик кариотипа | Хороший (норма, del(5q) del(20q) -Y) | Промежуточный (+8 хромосома, 2 аномалии и др.) | Плохой (аномалии 7 хромосомы, ≥ 3 аномалии) | — | — | |
| Цитопения (количество поражённых линий) | 0/1 | 2/3 | — | — | — | |
Сумма баллов, соответствующая высокому риску по IPSS (больше 2,5) складывается из мультилинейной дисплазии, плохого цитогенетического прогноза и высокого бластоза, на грани перехода в ОМЛ (срок трансформации в который в категории высокого риска составляет всего 2 месяца).
Категория промежуточного-2 риска также складывается из выраженного цитопенического синдрома и высокого, в пределах 10-20 % бластоза.
То, что в категории низкого риска медиана общей выживаемости ниже срока перехода в ОМЛ, объясняется меньшим сроком жизни больных с МДС, что отражает последствия осложнений цитопенического синдрома.[17]
| Сумма баллов | Риск по IPSS | Срок до перехода в ОМЛ у 25 % пациентов (лет) | Медиана общей выживаемости (лет) | % Пациентов |
|---|---|---|---|---|
| 0 | Низкий | 9,4 | 5,7 | 31 % |
| 0,5-1,0 | Промежуточный-1 | 3,3 | 3,5 | 39 % |
| 1,5-2,0 | Промежуточный-2 | 1,1 | 1,2 | 22 % |
| ≥ 2,5 | Высокий | 0,2 | 0,4 | 8 % |
| Баллы | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| Вид МДС по классификации ВОЗ | РА, РАКС, 5q- | РЦМД, РЦМД-КС | РАИБ1 | РАИБ2 |
| Кариотип | Хороший | Средний | Плохой | — |
| Потребность в гемотрансфузиях | Нет | Регулярная | — | — |
Кариотип:
- Хороший: нормальный, -Y, del 5q, del 20q
- Плохой: более 3х аномалий или аномалии 7 хромосомы
- Средний: все другие
Регулярные гемотрансфузии — переливание минимум 1 ЭМ каждые 8 недель в течение 4 месяцев.
| Группа риска | Баллы | Медиана выживаемости (мес) |
|---|---|---|
| Очень низкий | 0 | 136 |
| Низкий | 1 | 63 |
| Средний | 2 | 44 |
| Высокий | 3-4 | 19 |
| Очень высокий | 5-6 | 8 |
Не все пациенты с МДС нуждаются в терапии. Пациенты без анемического, геморрагического синдрома, инфекционных осложнений могут наблюдаться и не получать лечения (тактика «watch and wait»).
Выбор терапевтической тактики во многом определяется возрастом пациента, соматическим статусом, степенью риска по шкале IPSS, WPSS, наличием совместимого донора.
Можно выделить следующие направления терапии МДС:
- Сопроводительная терапия включает в себя переливание различных гемокомпонентов (эритроцитарной массы, тромбоконцентрата), терапию эритропоэтином, тромбопоэтином. У больных, часто получающих гемотрансфузии, развивается перегрузка организма железом. Железо обладает токсическим действием на различные ткани и органы, в первую очередь сердце, печень, поэтому такие пациенты должны получать препараты, связывающие железо, — хелаторы (десферал, эксиджад).
- Иммуносупрессивная терапия наиболее эффективна у пациентов с гипоклеточным костным мозгом, нормальным кариотипом и наличием HLA-DR15. Леналидомид, обладающий иммуномодулирующим и антиангиогенным действием, показал свою эффективность у трети пациентов с рефрактерной анемией (согласно критериям ВОЗ) и низким риском (по IPSS), а также у больных с 5q-синдромом. Эффективность лечения в данном случае весьма высока; 95 % больных достигают цитогенетической ремиссии.
- Аллогенная трансплантация гемопоэтических стволовых клеток от совместимых доноров является методом выбора у пациентов с миелодиспластическим синдромом.[19][20][21]
Пациентам с МДС моложе 65 лет, с хорошим соматическим статусом, при наличии HLA-совместимого донора показано проведение аллогенной трансплантации костного мозга, так как трансплантация является потенциально радикальным методом лечения МДС.
- Цитарабин, низкие дозы. Широко используются в России, да и во всей Европе, для лечения пациентов с МДС и ОМЛ, которым не подходит терапия методом ТКМ или применение интенсивной химиотерапии.
Мнения исследователей относительно целесообразности использования низкоинтенсивной терапии расходятся. Bowen D[22] считает, что нет оснований рекомендовать её рутинное использование при МДС: было выполнено 3 рандомизированных крупных исследования (141 пац.), которые показали, что применение низких доз цитарабина не увеличивает продолжительность жизни пациентов с МДС.[23] Вместе с тем, в более позднем исследовании у пациентов с ОМЛ и МДС высокого риска[24] было показано, что продолжительность жизни у больных, у которых применялся LDAC более, чем в 1 цикле, выше, чем при поддерживающей терапии.
Таким образом, необходимость в низкоинтенсивной терапии с доказанной эффективностью и лучшей переносимостью, чем LDAC, которая будет способствовать увеличению выживаемости пациентов с МДС высокого риска, остаётся актуальной.
- Высокодозная химиотерапия используется у больных с РАИБ с гипер- и нормоклеточным костным, при трансформации в ОМЛ. Пятилетняя выживаемость составляет около 18 %.
- Гипометилирующие препараты
Новые многообещающие терапевтические подходы, широко обсуждающиеся в последнее время, по поводу которых проводятся многочисленные клинические исследования, возникли в результате глубокого изучения биологии МДС. Среди них следует отметить ингибиторы метилирования ДНК (5-азацитидин, децитабин) и иммуномодулятор — леналидомид. 5-азацитидин обладает двойным механизмом действия. Он встраивается не только в молекулу ДНК, но и в молекулу РНК. В процессе метилирования ДНК гипометилирующие агенты ковалентно связываются с ДНК-метилтрансферазой, что приводит к реактивации генов, после чего восстанавливается дифференцировка гемопоэтических клеток-предшественников и нормальное кроветворение. Азацитидин, встраиваясь в РНК молекулу, тем самым понижает её количество в клетках, что приводит к цитостатическому эффекту вне зависимости от клеточной фазы. На основании результатов исследования 3 фазы AZA-001 — международное, мультицентровое, контролируемое, в параллельных группах, в котором пациенты МДС высокого риска/ОМЛ (ВОЗ критерии) сравнивались со стандартным лечением (сопроводительная терапия, интенсивная химиотерапия, низкие дозы цитарабина), азацитидин был зарегистрирован, в том числе и в РФ, для лечения этих больных. Было показано, что азацитидин в 2,5 раза увеличивает общую выживаемость.
- ↑ 1 2 Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Гематология / Под редак. О. А. Рукавицына. СПб.: 2007. С. 193—226
- ↑ Corey SJ, Minden MD, Barber DL et al. Myelodysplastic syndromes: the complexicity of stem-cell diseases. Cancer. Nature reviews. 2007. V.7; 118—129
- ↑ Pedersen-Bjergaard J, Pedersen M, Roulston D, Philip P. (1995) Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood. 86(9): 3542-3552.
- ↑ Greenberg P.L. Apoptosis and its role in the in the myelodysplastic syndromes; implications for disease natural history and treatment. Leuk res, 1998; 22: 1123—1136
- ↑ Onley HJ, Le Beau MM. Cytogenetic Diagnosis of Myelodysplastic syndromes. in book H.J.Deeg, D.T. Bowen, S.D.Gore, T.Haferlach, m.M.Le Beau and C.Niemeyr. Hematologic Malignancies: Myelodysplastic syndromes// Springler Berlin Heidelbery. 2006, P.55-79
- ↑ 1 2 NCCN Clinical Practice Guidelines in oncology. Myelodisplastic syndromes. V.1.2009 (англ.) : journal. Архивировано 31 октября 2010 года.
- ↑ Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, et al. (1982) Proposals for the classification of the myelodysplastic syndromes. Br.J Haematol. 51(2): 189—199.
- ↑ Brunning R, Bennett J, Flandrin G et al. Myelodysplastic Syndromes. In: Jaffe E, Harris N, Stein H et al, eds. WHO Classification of Tumours. Pathology and Genetics of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2001;61-73.
- ↑ Harris N, Jaffe E, Diebold J et al. WHO Classification of Neoplastic Diseases of the Hematopoietic and Lymphoid Tissues: Report of the Clinical Advisory Committee Meeting. J Clin Oncol 1999;17:3835-3849.
- ↑ Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002;100:2292-2302.
- ↑ Bruning RD, Orazi A., Germing U. Et al., 2008. WHO classification of tumors of hematopoietic and lymphoid tissues. Chapter 5, pp 88-107
- ↑ Hollstrem Lindberg E., Cazzola M. The role of JAK2 mutations in RARS and other MDS. 2008. Hematology, 52-59
- ↑ Greenberg PL, Baer M, Bennett J et al. NCCN Practice Guidelines for Myelodysplastic Syndromes, Version1, 2001, In "The Complete Library of NCCN Guidelines [CD-ROM], " Rockledge, PA.
- ↑ Cheson BD, Bennett JM, Kantarjian H et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 2000;96:3671-3674.
- ↑ 1 2 Greenberg P., Cox c., Le Beau MM et al., International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89:2079-2088
- ↑ Malcovati L., Germing U., Kuendgen A. Et al. Time-dependant prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25: 3503-10
- ↑ A WHO Classification-Based Prognostic Scoring System (WPSS) for Predicting Survival in Myelodysplastic Syndromes. Luca Malcovati et al. ASH Annual Meeting Abstracts 2005 106: Abstract 788
- ↑ Scott BL, Sandmaier BM, Storer B et al. Myeloablative vs nonmyeloablative allogeneic transplantation for patients with myelodysplastic syndrome or acute myelogenous leukemia with multilineage dysplasia: a retrospective analysis. Leukemia 2006;20:128-135.
- ↑ Wallen H, Gooley TA, Deeg HJ et al. Ablative allogeneic hematopoietic cell transplantation in adults 60 years of age and older. J Clin Oncol 2005;23:3439-3446.
- ↑ Demuynck H, Verhoef GE, Zachee P et al. Treatment of patients with MDS with allogeneic bone marrow transplantation from genotypically similar HLA-identical sibling and alternative donors. Bone Marrow Transplant 1996;17:745-751.
- ↑ Bowen D. Is traditional low dose chemotherapy (cytarabine/melphalan) still on option? Leukemia Research, Volume 31, Supplement 1, May 2007, Page S19
- ↑ Miller K.B. et al. The evaluation of low-dose cytarabine in the treatment of myelodisplastic syndromes: a phase III intergroup study. Annals of hematology, 1992; 65: 162—168.
- ↑ Burnett A.K., Milligan D., Prentice A.G. et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007. 109: 1114—1124
Образовательный видеоролик в помощь пациентам с миелодиспластическим синдромом
Миелодиспластический синдром: развитие, лечение, прогноз
Миелодиспластический синдром (МДС) — тяжелое гематологическое заболевание, которое относится к группе онкопатологий и плохо поддается терапии. В основе недуга лежит нарушение процесса воспроизведения клеток крови: их развития и деления. В результате подобных аномалий образуются онкологические структуры, и формируются незрелые бласты. Постепенно количество нормально функционирующих, зрелых клеток в организме уменьшается. Данный синдром называют «дремлющим лейкозом» из-за скопления в крови бластных клеток.
Костный мозг — важный кроветворный орган, в котором происходят процессы образования, развития и созревания клеток крови, то есть осуществляется гемопоэз. Этот орган также принимает участие в иммунопоэзе — процессе созревания иммунокомпетентных клеток. У взрослого человека в костном мозге содержатся незрелые, недифференцированные и низкодифференцированные клетки – стволовые.
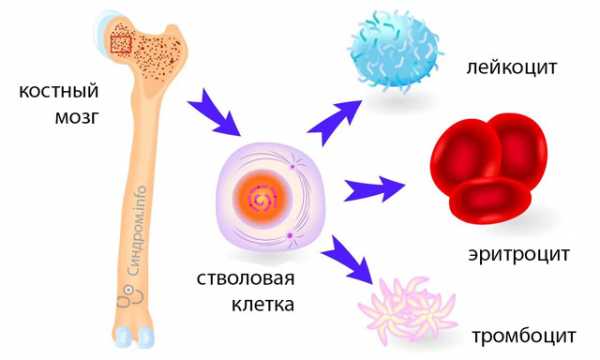
Большинство заболеваний костного мозга обусловлены мутацией стволовой клетки крови и нарушением ее дифференциации. МДС не является исключением. Расстройство кроветворения приводит к развитию острого лейкоза. Причина первичного МДС неизвестна. Мутагенные факторы оказывают негативное воздействие на стволовую клетку крови, что приводит к нарушению ДНК и выработке в костном мозге аномальных клеток, постепенно вытесняющих нормальные. Вторичный синдром развивается в результате длительного лечения цитостатиками, при частом контактировании с химическими веществами, в следствии облучения. Заболевание чаще развивается у пожилых лиц старше 60 лет, чаще у мужчин. Раньше среди детей синдром практически не встречался. В настоящее время недуг «помолодел». Все чаще случаи МДС наблюдаются у больных среднего возраста, что связано с экологическими проблемами крупных городов. Миелодиспластический синдром имеет код по МКБ-10 D46.
Цитопения — клиническое проявление патологий системы кроветворения. Симптоматика недуга определяется поражением определенной клеточной линии. У больных возникает слабость, утомляемость, бледность, головокружение, лихорадка, кровоточивость, кровоизлияния. Специфические признаки при этом отсутствуют. Диагностика патологии основывается на результатах гемограммы и гистологического исследования биоптата костного мозга. Лечение заключается в переливании основных компонентов крови, проведении химиотерапии, иммуносупрессивной терапии и пересадке костного мозга.
цитопения с нарушением созревания клеток крови по нескольким росткам
Эффективное лечение МДС – одна из самых сложных проблем современной медицины. Его проводят специалисты в области онкогематологии. Синдром в запущенных случаях приводит к онкологии. Но так происходит не всегда. Легкие формы недуга типа рефракционной анемии обычно не заканчиваются формированием рака. Недостаток клеток крови приводит к анемии, кровоточивости, сердечной дисфункции, увеличению риска развития инфекционных заболеваний. Прогноз МДС определяется особенностями течения патологического процесса, своевременностью диагностических и общетерапевтических мероприятий. Своевременная терапия – единственный реальный шанс сохранить и продлить жизнь больных.
Этиология и патогенез
Гемопоэз – процесс кроветворения, который заключается в образовании и созревании клеток крови. Он происходит непрерывно, что связано с коротким сроком жизни клеток: от нескольких дней до 3-4 месяцев. Ежедневно в живом организме синтезируется огромное количество новых кровяных телец из клеток-предшественников. В процессе миелопоэза образуются миелоидные клетки – эритроцитарные, лейкоцитарные и тромбоцитарные клеточные элементы. Под воздействием негативных экзогенных и эндогенных факторов в костном мозге происходят патологические изменения, возникает расстройство кроветворения.
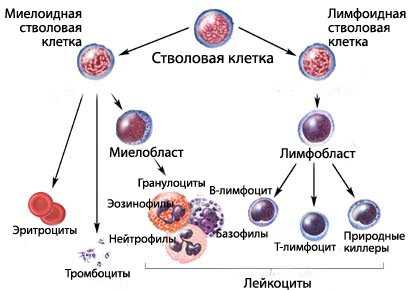
Этиология и патогенез МДС в настоящее время полностью не изучены. Ученые установили факторы, провоцирующие развитие патологии:
- загрязнения окружающей среды,
- радиоактивное излучение,
- табакокурение,
- опасные и вредные производственные факторы,
- контакты с агрессивными веществами,
- длительное проведение иммуносупрессивной терапии,
- врожденные генетические заболевания.
Первичный или идиопатический синдром — недуг невыясненной этиологии, который развивается в 80% случаев у лиц в возрасте 60-65 лет.
Вторичный синдром обусловлен воздействием на организм химиотерапевтических препаратов или лучевой терапии. Эта форма обычно развивается у молодых людей, быстро прогрессирует, отличается высокой устойчивостью к лечению и максимальным риском развития острого лейкоза.
В костном мозге вырабатываются все клеточные элементы крови. Там они находятся в незрелом состоянии, то есть являются предшественниками зрелых форм. По мере необходимости каждая из них превращается в полноценные клетки и выполняет жизненно важные функции, от которых зависит процесс дыхания, гемостаз, иммунная защита. При МДС стволовые клетки погибают до выхода в кровяное русло и не достигают своей функциональной зрелости. Это приводит к дефициту нормальных клеточных форм в крови и нарушению их функций, связанному с клеточной дисплазией.
МДС часто называют тлеющей лейкемией или предлейкозом, обусловленном генной мутацией стволовых клеток. Клональная пролиферация эритроидных, миелоидных и мегакариоцитарных форм приводит к неэффективному гемопоэзу и панцитопении. В костном мозге и крови происходят характерные морфологические изменения, обусловленные аномальной клеточной продукцией. У больных увеличивается печень и селезенка. Нестабильность синдрома обусловлена тенденцией к переходу в острый миелобластный лейкоз.
Симптоматические проявления
МДС не имеет специфической симптоматики. Его клинические проявления определяются степенью тяжести и формой недуга.
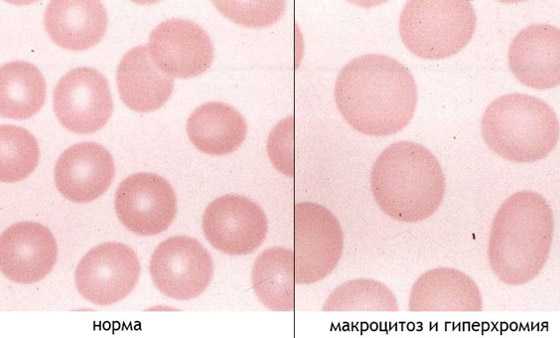 Анемический синдром — постоянный и обязательный признак патологии. Для него характерны гиперхромия и макроцитоз. Большой размер эритроцитов и их интенсивное окрашивание, зависящее от повышенного содержания гемоглобина, – признаки анемии при МДС и остром лейкозе. При анемии больные быстро утомляются, плохо переносят физические нагрузки, жалуются на головокружение, одышку, боль в груди, костях и суставах, невозможность сосредоточится. Их кожа становится бледной, ухудшается аппетит, снижается вес и работоспособность, возникает нервозность, цефалгия, дрожь в теле, шум в ушах, сонливость, тахикардия, обмороки. Плохо переносят анемию престарелые больные, а также лица с сердечно-легочной патологией. У них могут развиться тяжелые последствия – стенокардия, инфаркт миокарда, аритмии.
Анемический синдром — постоянный и обязательный признак патологии. Для него характерны гиперхромия и макроцитоз. Большой размер эритроцитов и их интенсивное окрашивание, зависящее от повышенного содержания гемоглобина, – признаки анемии при МДС и остром лейкозе. При анемии больные быстро утомляются, плохо переносят физические нагрузки, жалуются на головокружение, одышку, боль в груди, костях и суставах, невозможность сосредоточится. Их кожа становится бледной, ухудшается аппетит, снижается вес и работоспособность, возникает нервозность, цефалгия, дрожь в теле, шум в ушах, сонливость, тахикардия, обмороки. Плохо переносят анемию престарелые больные, а также лица с сердечно-легочной патологией. У них могут развиться тяжелые последствия – стенокардия, инфаркт миокарда, аритмии.- Нейтропения характеризуется лихорадкой, снижением сопротивляемости организма к патогенным биологическим агентам, частым развитием инфекционных заболеваний бактериальной и вирусной этиологии. У больных повышается температура тела, потливость, возникает слабость, увеличиваются лимфоузлы. Сепсис и пневмония у таких пациентов часто заканчиваются летальным исходом.
- При тромбоцитопении кровоточат десна, появляются гематомы и петехии, часто течет кровь из носа, возникают длительные кровотечения после мелких хирургических вмешательств и различных инвазивных манипуляций. Возможно развитие внутренних кровотечений, меноррагий, кровоизлияний в головной мозг. Массивная потеря крови часто становится причиной смерти пациентов.
- У больных возникает лимфаденит, гепатомегалия, спленомегалия, специфическое поражение кожи — лейкемиды.
МДС долгое время может протекать бессимптомно или иметь стертое течение. Больные часто не обращают внимание на слабовыраженные клинические проявления и не посещают своевременно врача. Обычно МДС обнаруживают случайно во время проведения очередного медосмотра.
Диагностика
Диагноз МДС ставят после проведения лабораторного исследования периферической крови и гистологического исследования биоптата костного мозга. Специалисты изучают образ жизни больного, его анамнез, наличие профессиональных вредностей.
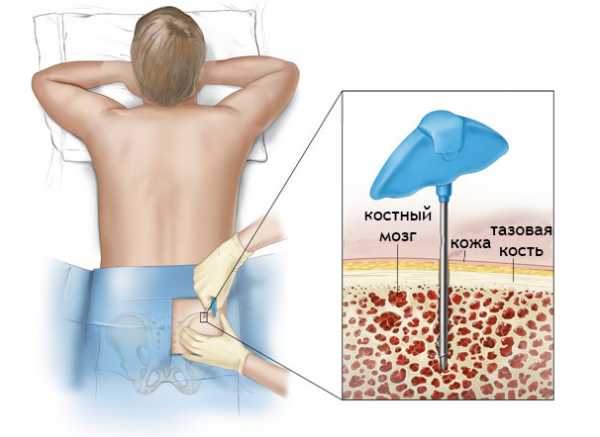
наиболее достоверный диагностический метод – трепанбиопсия костного мозга
Диагностические методы при МДС:
- гемограмма — анемия, лейкопения, нейтропения, моноцитоз; панцитопения – абсолютное показание для цитологического исследования костного мозга;
- биохимия крови – определение уровня железа, фолиевой кислоты, эритропоэтина, ЛДГ и АСТ, АЛТ, щелочной фосфатазы, мочевины;
- иммунограмма – специальный комплексный анализ, позволяющий определить, в каком состоянии находится иммунная система;
- гистология костного мозга выявляет деструкцию ткани, очаги поражения, наличие аномальных клеток, дисбаланс кроветворной и жировой ткани, гиперплазию всех ростков кроветворения, признаки дисплазии клеток;
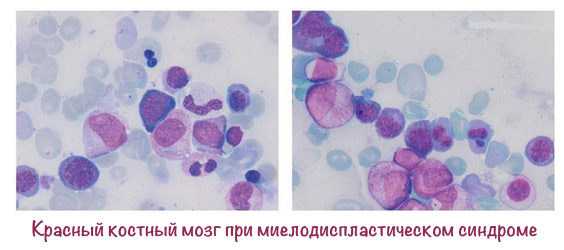
- цитохимическое исследование – нарушение обмена микроэлементов и витаминов: щелочной фосфатазы в лейкоцитах, миелопероксидазы, железа;
- цитогенетический анализ – выявление хромосомных аномалий;
- дополнительные инструментальные исследования, позволяющие оценить состояние внутренних органов — УЗИ, КТ и МРТ.
Только после полноценной диагностики и постановки правильного диагноза можно переходить к лечению недуга.
Лечение
Интенсивное лечение МДС заключается в применении целого комплекса мероприятий. В тяжелых случаях медикаментозную терапию проводят в условиях стационара. Больные с более легкими формами синдрома лечатся амбулаторно или на дневном стационаре. Основными среди общетерапевтических мероприятий являются химиотерапия и иммуносупрессивные методики. Трансплантация костного мозга проводится при тяжелом течении болезни и повышает шансы больных на выздоровление.
Лечение МДС проводится с целю нормализации показателей периферической крови, устранения симптомов патологии, предупреждения трансформации недуга в острый лейкоз, улучшения и продления жизни больных.
Симптоматическая терапия направлена на устранение клинических проявлений синдрома и сопутствующих заболеваний, осложняющих течение основного недуга.
- Внутривенное капельное введение кровяных ингредиентов – тромбоконцентрата или эритроцитарной массы. Тромбоцитарную массу переливают редко.
- Для профилактики гемосидероза – «Дисферал».
- Иммуносупрессоры – «Леналидомид», антитимоцитарный и антилимфоцитарный глобулин, «Циклоспорин А», комбинации глюкокортикоидов.
- Химиотерапевтические средства – «Цитарабин», «Дакоген», «Мельфалан».
- Препараты-стимуляторы эритропоэза — железосодержащие препараты: «Ферроплекс», «Фенюльс», «Сорбифер дурулес»; препараты витаминов: «Цианкобаламин», «Фолиевая кислота»; анаболики-стероиды: «Анаполон», «Нандролон»; препараты эритропоэтина: «Эральфон», «Эпокомб»
- Стимуляторы лейкопоэза – «Нейпоген», «Лейкоген», «Метилурацил», «Интерлейкин».
- Ингибирование апоптоза – естественной гибели клеток – «Сандиммун», «Весаноид».
- Ингибиторы развития кровеносных сосудов – «Талидамид», «Ревлимид».
- Гипометилирующие средства – «Азацитидин».
- При развитии инфекционных осложнений — антибиотики и антимикотики.
Схема лечения и дозировка препаратов зависят от возраста пациента, степени тяжести заболевания и общего состояния здоровья. Эффективность медикаментозной терапии достаточно низкая и непродолжительная. Единственный способ спасти больного — выполнить пересадку костного мозга. В тяжелых случаях также проводят трансплантацию стволовых клеток. Несмотря на свою эффективность, эти способы лечения имеют много недостатков: являются дорогостоящими, имеют высокую вероятность отторжения трансплантата, требуют дополнительной подготовки пациента к операции, вызывают трудности в поиске подходящего донора.
В настоящее время развитие генной инженерии и культивирование клеток крови достигли нового уровня. С их помощью процесс кроветворения можно регулировать. Специалисты определяют сколько клеток недовырабатывается индивидуально у каждого больного, а затем переходят непосредственно к лечению.
С помощью любого из вышеперечисленных методов можно добиться полной ремиссии синдрома.
Профилактика
Специфической профилактики синдрома не существует. Профилактические мероприятия, не допускающие ухудшения состояния больных и предупреждающие трансформацию синдрома в лейкоз:
- укрепление иммунитета,
- сбалансированное питание,
- поддержание гемоглобина на оптимальном уровне,
- частые прогулки на свежем воздухе,
- своевременное обращение к врачу при появлении первых признаков синдрома,
- периодическая сдача анализов и прохождение необходимых исследований,
- гигиена кожи,
- защита от контактов с химическими веществами,
- защита от радиации,
- ограничение активной физической нагрузки,
- своевременное лечение простудных и инфекционных заболеваний.
Прогноз
Прогноз МДС неоднозначный. Он зависит от тяжести патологии и своевременности лечения. Продолжительность жизни при легких формах синдрома составляет 15 лет, при наличии тяжелого течения недуга она не превышает 10 месяцев. При отсутствии или неэффективности лечения МДС трансформируется в острый лейкоз. Адекватная терапия обеспечивает максимальное продление жизни. Наблюдение за больными, имеющими стертую клиническую картину и относительно благоприятное течение недуга, осуществляют постоянно, даже в период стабильных показателей крови и костного мозга.
У пожилых людей синдром протекает особенно тяжело и плохо лечится. Это связано с наличием у них хронических заболеваний и подавлением иммунитета. Их организм не справляется, и процесс выздоровления затягивается.
Видео: базовая информация о миелодиспластическом синдроме
Миелодиспластический синдром - причины, симптомы, диагностика и лечение, прогноз
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Общие сведения
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Миелодиспластический синдром
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Миелодиспластические синдромы | Фонд «Подари жизнь»
Суть болезни
Миелодиспластические синдромы (МДС) – группа заболеваний, которые характеризуются нарушениями кроветворения миелоидной линии. В результате этих нарушений возможность выработки зрелых клеток крови частично сохраняется, но наблюдается дефицит тех или иных их видов, а сами клетки при этом изменены и плохо функционируют.
У значительной части больных МДС через некоторый промежуток времени, обычно от нескольких месяцев до нескольких лет, развивается острый миелоидный лейкоз.
МДС в обиходе иногда называют «предлейкемией», ранее применялись также термины «малопроцентный лейкоз», «тлеющий лейкоз» или «дремлющий лейкоз». Это связано с содержанием бластных клеток в костном мозге: если их более 20% (согласно классификации Всемирной организации здравоохранения) или более 30% (согласно франко-американо-британской классификации FAB), то речь уже идет о миелоидном лейкозе, если же их уровень ниже порогового значения, то может быть диагностирован МДС.
Под термином «миелодиспластический синдром» в настоящее время подразумевается целая группа заболеваний, различающихся по частоте встречаемости, клиническим проявлениям, а также по вероятности и ожидаемым срокам трансформации в лейкоз. Специалисты используют две классификации МДС: франко-американо-британскую (FAB) и классификацию Всемирной организации здравоохранения (ВОЗ). Рассмотрим классификацию FAB как более простую:
- Рефрактерная анемия (РА). Термин «рефрактерная» здесь означает, что анемия не поддается лечению препаратами железа и витаминами. В костном мозге менее 5% миелобластов, аномалии в основном касаются предшественников эритроцитов.
- Рефрактерная анемия с кольцевыми сидеробластами (РАКС): миелобластов в костном мозге менее 5%, но не менее 15% предшественников эритроцитов представлены особыми аномальными клетками – так называемыми кольцевыми сидеробластами. Это клетки с кольцеобразными «отложениями» железа, которые не могут обеспечивать эффективный транспорт кислорода.
- Рефрактерная анемия с избытком бластов (РАИБ): миелобластов в костном мозге 5–20%. В классификации ВОЗ дополнительно подразделяется на РАИБ-I (5–9% бластов) и РАИБ-II (10-19% бластов).
- Рефрактерная анемия с избытком бластов на стадии трансформации (РАИБ-T): миелобластов 21–30% (по классификации ВОЗ это уже острый миелоидный лейкоз).
- Хронический миеломоноцитарный лейкоз, ХММЛ (по классификации ВОЗ относится к миелодиспластическим-миелопролиферативным заболеваниям).
Частота встречаемости, факторы риска
Этопозид — Википедия
Этопозид — ингибитор топоизомеразы II. Является полусинтетическим производным подофиллотоксина. Оказывает цитотоксическое (не путать с цитостатическое) действие за счет повреждения ДНК.
Механизм действия связан с ингибированием топоизомеразы II. Угнетает митоз, блокирует клетки в S-G2-интерфазе клеточного цикла, в более высоких дозах действует в фазе G2. Цитотоксическое действие в отношении нормальных здоровых клеток наблюдается только при использовании в высоких дозах.
При приеме внутрь этопозид всасывается из ЖКТ. Биодоступность в среднем составляет 50 %. Распределение в спинномозговой жидкости низкое и вариабельное; концентрация в нормальной легочной ткани выше, чем при наличии метастазов в легких; определяется близкий уровень концентраций в тканях первичных опухолей и в нормальных тканях миометрия. Отмечена прямая корреляция между коэффициентом связывания этопозида и уровнем альбумина в плазме крови здоровых людей и пациентов с раковыми заболеваниями. Метаболизируется в печени. Конечный T1/2 в среднем составляет 7 ч. Выводится почками — 44-60 %, с фекалиями — до 16 %, с желчью — 6 % и менее.
Герминогенные опухоли (опухоли яичка, хориокарцинома), рак яичников, мелкоклеточный и немелкоклеточный рак легкого, лимфогранулематоз, неходжкинские лимфомы, рак желудка (для монотерапии и в составе комбинированной терапии), саркома Юинга, саркома Капоши, нейробластома, рак молочной железы (с метастазами в печень, в плевру), острый нелимфобластный лейкоз, мезотелиома.
Устанавливают индивидуально, в зависимости от показаний и стадии заболевания, состояния системы кроветворения, схемы противоопухолевой терапии.
Со стороны системы кроветворения: лейкопения, анемия; реже — тромбоцитопения. Со стороны пищеварительной системы: тошнота, рвота; редко — анорексия, мукозиты, диарея; при применении высоких доз — токсические реакции со стороны печени. Со стороны ЦНС и периферической нервной системы: сонливость, повышенная утомляемость; поражение периферической нервной системы. Со стороны обмена веществ: гиперурикемия; при применении высоких доз — метаболический ацидоз. Со стороны сердечно-сосудистой системы: тахикардия, артериальная гипотензия. Со стороны репродуктивной системы: азооспермия, аменорея. Аллергические реакции: озноб, лихорадка, бронхоспазм. Дерматологические реакции: алопеция.
Выраженная миелодепрессия, выраженные нарушения функции печени и почек, беременность, детский возраст до 2 лет, повышенная чувствительность к подофиллину или его производным.
Этопозид противопоказан при беременности. При необходимости применения в период лактации следует прекратить грудное вскармливание.
Во время лечения и в течение трёх месяцев после его окончания пациенткам детородного возраста необходимо применять эффективные методы контрацепции.
В экспериментальных исследованиях установлено, что этопозид оказывает тератогенное и эмбриотоксическое действие.
С осторожностью применяют у пациентов с предшествующей лучевой или химиотерапией, с ветряной оспой, опоясывающим герпесом, при инфекционных поражениях слизистых оболочек, при нарушениях сердечного ритма, при повышенном риске развития инфаркта миокарда, нарушении функции печени, заболеваниях нервной системы (эпилепсия), хроническом алкоголизме, у детей старше двух лет.
При нарушении функции почек дозу уменьшают в соответствии со значениями КК.
Перед началом и на фоне проводимой терапии следует контролировать картину периферической крови.
Не рекомендуют проводить вакцинацию пациентов и членов их семей.
В экспериментальных исследованиях установлено, что этопозид оказывает мутагенное действие.
Влияние на способность к вождению автотранспорта и управлению механизмами: во время лечения следует воздерживаться от занятий, требующих повышенного внимания и быстрых психомоторных реакций.
Этопозид включен в Перечень Жизненно необходимых и важнейших лекарственных препаратов.
При одновременном применении с другими препаратами, вызывающими миелодепрессию возможно аддитивное угнетение функции костного мозга.
При одновременном применении с цисплатином возможно уменьшение клиренса этопозида и увеличение его токсичности.
Циклоспорин в высоких дозах может уменьшать клиренс этопозида и увеличить продолжительность его действия, при этом возможно усиление лейкопении.
виды, симптомы, лечение, причины и прогноз жизни
Цитостатические лекарственные средства применяются для сдерживания и уничтожения злокачественных клеток. Однако после химиотерапии такими препаратами проявляется негативный побочный эффект: ухудшение работы костного мозга и последующее уменьшение в организме кровяных частиц, называемое цитопенией. Отличие патологии выделяется из её названия (греч. cyto – клетка, penia – недостаток). Обычно наблюдается изолированная нехватка лейкоцитов и тромбоцитов, реже – эритроцитов.
Причины возникновения и развития цитопении
Проявление заболевания выражается в двух факторах: разрушение кровяных телец в сосудах, а также ухудшение синтеза новых клеток.
Не считая воздействия на организм ряда медикаментов, причинами возникновения цитопении являются:
- Анемия (в том числе постцитостатическая), вызванная нехваткой железа и витаминов группы B.
- Инфекционные заболевания (туберкулёз, сифилис, гепатит, пневмония)
- Высокая скорость кровяного цикла.
- Миелодиспластический синдром и причины его образования (негативная генетика, вредное производство, курение, радиация).
- Онкологии, вызывающие ухудшение кровяного цикла: все виды лейкоза, метастазы костного мозга, миелома, лимфосаркома.
- Депрессия, частые стрессы, проявления шизофрении и другие ухудшения работы нервной системы.

- Дифференциация стволовых клеток в костном мозге, нарушающая его деятельность.
- При увеличении селезёнки – секвестрация в ней кровяных тел.
- Наследственная нейтропения (замедление роста и снижение концентрации отдельного вида белых тел в крови).
- Уничтожение эритроцитов антителами (аутоиммунная гемолитическая анемия).
- Цитомегалия и мононуклеоз, провоцирующие проблемы с балансом гормона роста и приводящие к остановке роста клеток крови.
- Пароксизмальная ночная гемоглобинурия (в кровеносных сосудах появляются склонные к гемолизу клоны эритроцитов).
Следствие развития цитопении – характерные для неё клинические признаки:
- уменьшение синтеза клеток крови и замедление их выхода;
- ускорение цикла кровообращения;
- поглощение и перераспределение кровяных тел в сосудах.
Виды цитопении и их характеристики
Цитопения – это ощутимый для организма дефицит клеток крови в организме. Болезнь обнаруживает разновидности: названия и особенности обусловлены нехваткой конкретного компонента системы кровообращения.
Двух-, трёхростковая, рефрактерная цитопения, панцитопения считаются симптомами миелодиспластического синдрома (МДС) – неспособности костного мозга воспроизводить клетки крови. Группа заболеваний МДС располагает кодом по МКБ-10 D46.
Эритроцитопения
Нехватка красных кровяных тел. Патология вынуждает уменьшать количество гемоглобина и кислорода в организме, приводящее к утомлению и ухудшению состояния больного. По признакам патология характерна типичному оголоданию человека: озноб, слабое внимание, потеря массы, головокружение. Все видимые покровы портятся (растрескивание губ, бледность или пожелтение кожи, кариес и рыхлые дёсны на зубах).
Видимые проявления тромбоцитопении
Лейкопения
Лейкопения – пониженный уровень лейкоцитов в системе кровообращения. Уменьшение их количества негативно сказывается на иммунной системе человека: сопротивление болезням слабеет, даже незначительные недуги протекают в тяжёлой форме. Сопровождается частыми инфекционными заболеваниями, недомоганием и высокой температурой тела. На теле появляются прыщи, болячки, фурункулы, нагноения.
Тромбоцитопения
Тромбоцитопения представляет собой изолированный дефицит тромбоцитов в цикле кровообращения. Имеет код по МКБ-10 D69. Внешне распознаётся частыми кровотечениями из носа, кровохарканием, черным калом (кровоизлияния в кишечнике). Телесные повреждения (и сильные, и слабые) вызывают синяки. Причины – ухудшение свёртываемости и меньшая вязкость крови: как итог, кровотечение (как внутреннее, так и наружное) занимает больше времени для остановки.
Другие виды
Пониженное содержание всех видов составляющих цикла кровообращения приводит к панцитопении.
Угнетение работы нескольких ростков кроветворения означает заболевание двухростковой или трёхростковой цитопенией. Также возможна рефрактерная цитопения – поражение всех ростков: нехватка гемоглобина, тромбоцитов и лейкоцитов. Тяжёлым видам недуга подвержены люди старше 70 лет.
Диагностика
Обнаружить и искоренить сразу недуг не получится: на первых порах заметных признаков нет. Количество элементов крови уменьшается постепенно: к плавным переменам организм стремится приспосабливаться. Симптомы проявляются только при длительном развитии патологии.
Электронная фотография эритроцита крови
Диагностика цитопении
Вероятное развитие цитопении возможно отследить при долгом наблюдении ряда общих симптомов: по частой заболеваемости, утомлению, недомоганиям, ломким ногтям, выпадению волос.
Точное определение наличия и разновидности патологии устанавливается специалистами. Диагностика заболевания проходит в ряд последовательных шагов:
- Началом послужит беседа больного с врачами. Затем, собрав необходимые сведения (анамнез, вероятность наследственных патологий) и проведя полный клинический осмотр, устанавливается характер заболевания.
- Следующий шаг – внутренний осмотр. Включает общий и биохимический анализ крови и мочи. Проводится иммунологическая и серологическая экспертиза (на уровень сопротивляемости организма и стадию развития уже имеющейся болезни).
- Забор пункции костного мозга, анализ полученных тканей.
- Выведение миелограммы (результатов анализа состояния костного мозга): изучение хода кроветворения.
- УЗИ брюшной полости (проверка на увеличение органов), МРТ и ПЭТ при необходимости.
Диагностика МДС
Обнаруживается и устанавливается диагноз МДС после гистологического и цитологического изучения поражённого органа.
Двухростковая цитопения – синдром рефрактерной цитопении с мультилинейной дисплазией (РЦМД): 10% клеток с нарушенным развитием, дефект двух или трёх ростков кроветворения.
Цитопения конкретного вида клеток – неклассифицированный МДС (с одноростковой дисплазией и отсутствием бластов) или рефракторная анемия (при наличии бластов).
Панцитопения вкупе с повышенным числом кольцевых сидеробластов (свыше 15%) говорит о РЦМД с кольцевыми сидеробластами.
Лечение цитопении
В случае подтверждения заболевания врачом-гематологом проводится лечение. Оно требует стационарных условий, а также регулярно сдавать кровь на анализ. Важно помнить: указанная болезнь – не самостоятельный недуг, а сопровождение другой патологии. Курс, нацеленный только на применении мер против цитопении, будет бесполезным.
Общее лечение заболевания
Обычное лечение (на предмет сниженного иммунитета) предусматривает употребление пациентом гормональных кортикостероидов и глюкокортикостероидов. Также гематологами выявляется нехватка минералов и иных компонентов крови: составляются курсы употребления витаминов и правильного питания, сравнение состояния пациента до, во время и после терапии. При острой необходимости допускается переливание крови. Результат терапии непредсказуем: возможен как положительный, так и негативный итог. Во время лечения процедуры и препараты подбираются с учётом особенностей патологии.
Лечить пациента стандартным курсом достаточно, если основной болезнью стала лёгкая или средняя степень анемии: восполняется недостаток железа, белков и витамина В12.
Цитопения обнаруживается не только у взрослых, но и у детей всех возрастов. Их комплекс лечения предполагает принятие Цитозара – предусматривается как дифференциальная, так и комбинированная терапия. Универсальность препарата делает его ключевым средством в достижении ремиссии раковых опухолей.
Специфическое лечение цитопении
По сравнению с консервативной терапией, к углублённому варианту лечения недуга прибегают, если заболевание-источник признан тяжёлой патологией.
При лейкозе, миеломе, метастазах костного мозга и иных онкологических заболеваниях в развитой форме стационарного лечения и насыщения организма становится недостаточно. Если применение цитозара также не привело к желаемым результатам, проводится хирургическое вмешательство: выполняется пересадка костного мозга. Эта операция считается наиболее действенным способом избавления от цитопении. Другой возможной процедурой (при соответствующих медицинских показаниях) считается удаление селезёнки.
После выполненных хирургических процедур назначается симптоматическое лечение. От правильно организованной диеты, восполняющей послеоперационную бедность организма питательными веществами, витаминами и минералами, и распределения физических нагрузок зависит восстановление организма и возвращение полноценной жизнедеятельности человека. Симптомы проходят, состояние нормализуется.
Профилактика и прогноз жизни
Методы, предупреждающие развитие цитопении на ранних сроках, отсутствуют: из-за адаптации организма начальную стадию невозможно распознать. Цель профилактических мер – уменьшить число обострений.
Предупреждающие мероприятия
Для профилактики переболевшему патологией нужно регулярно сдавать анализ крови и проходить обследование у врача-гематолога. Норма посещений – не реже 1 раза в полгода (или каждые 4 месяца, если риск рецидива велик).
Выздоровевший пациент для профилактики цитопении должен соблюдать каждый элемент здорового образа жизни:

- Здоровое питание: обогащение рациона свежими фруктами, ягодами, орехами, злаками, нежирным мясом и молоком, избегание жареной и жирной еды.
- Занятия лечебной физкультурой, двигательная активность: развивается кровообращение, улучшается иммунитет, стабилизируется работа сердца и лёгких.
- Полный отказ от вредных привычек: курения, алкоголя, переедания.
- Следование рекомендациям лечащего врача, регулярное употребление прописанных препаратов.
Сроки жизни после хирургической и химической терапии
Прогноз жизни и возможность полноценной жизнедеятельности зависит от общего физического состояния пациента, его возраста и заболевания, послужившего причиной развития цитопении. Практика показывает: после химиотерапии и остальных необходимых мер 4 из 5 переболевших гарантировано восстановление до нормы и пятилетний срок жизни.
При лейкозе в ранней стадии, анемии, инфекционных заболеваниях в случае их успешного преодоления обычно даётся положительный прогноз жизни.
Лечение миелодиспластического синдрома в тяжёлой форме зависит от своевременного переливания крови и трансплантации костного мозга (в особых случаях – стволовых клеток). Но, учитывая, что 80% больных – люди старше 60 лет (с ожидаемым сроком жизни до 12 лет), вопрос о применении хирургических мер почти не поднимается. Основной метод – лечение низкой интенсивности (переливание эритроцитов, белковое питание, химиотерапия) и поддерживающие меры для улучшения состояния и посильного продления существования.
В сравнении с довольно «лёгкими» патологиями, на поздних сроках онкологических недугов и метастазов в системе кровообращения также применяются лишь общие лечебные процедуры по поддержанию внешнего качественного состояния здоровья пациента. Неизлечимые патологии неизбежно ведут к смерти больного.
Миелодиспластический синдром переходит в рак?
К возникновению лейкоза в острой форме, диспластическим отклонениям в костном мозге может привести миелодиспластический синдром. Патология чаще поражает лиц мужского пола после 60 лет, а у детей диагностируется лишь в 5% случаев.
Что такое миелодиспластический синдром?
Миелодиспластический синдром или МДС – группа заболеваний онкологического типа, которые характеризуются нарушением воспроизведения нескольких или одного вида клеток крови. При патологии стволовые клетки не могут правильно развиваться и делиться, они превращаются в онкологические структуры или формируют бласт незрелых клеток. Нормально функционировать в человеческом организме способны только зрелые клетки, количество которых при МДС уменьшается. Такое состояние в медицине называют цитопенией.
По типу кровяных клеток, цитопению подразделяют на 3 вида:
- Анемия – при недостатке гемоглобина в крови.
- Нейтропения – при дефиците лейкоцитов в организме.
- Тромбоцитопения – при нехватке тромбоцитов.
Чем опасен миелодиспластический синдром?
Из-за недостаточного количества клеток крови у пациентов развивается анемия, сердечная недостаточность, увеличивается риск заражения инфекционными возбудителями. МСД, связанный с анемией, становится причиной быстрой утомляемости и головокружений у больного. Этот вид патологии считается наиболее легким, среди всех имеющихся типов.
Недостаток лейкоцитов в крови сказывается на работе иммунной системы – человек становится уязвимым перед инфекционными заболеваниями. Нарушение функционирования защитных сил организма приводит к различным осложнениям – от стоматита до пневмонии.
При тромбоцитопении нарушается сворачиваемость крови, в результате чего любые травмы и порезы могут привести к обильной кровопотере. Серьезные травмы при патологии становятся причиной летального исхода.
Миелодиспластический синдром и онкология
Миелодиспластический синдром и рак крови взаимосвязаны. МДС в запущенных стадиях может привести к онкологии. Но синдром не обязательно перерастает в рак. При легких типах заболевания, например, при рефракторной анемии, не возникает перерождения незрелых клеток в злокачественные.
Вторичные формы МСД встречаются среди больных раком. Причиной этому служат препараты, применяемые во время химиотерапии. В данном случае заболевание протекает в острой форме и трудно поддается медикаментозной терапии.
Как часто встречается МДС?
По причинам возникновения МДС подразделяют на первичный и вторичный тип. Идиопатический тип патологии диагностируется в большинстве случаев (80-90%) у пациентов старше 60 лет. Вторичный тип встречается в 10% случаев и может быть диагностирован в любом возрасте.
В последнее время заболевание все чаще встречается у пациентов трудоспособного возраста. Считается, что причиной «омоложения» МДС является неблагоприятная экологическая обстановка.
Причины и группы риска
Точные причины проблемы не изучены. К факторам, способствующим развитию заболевания, относят:
- наследственные генные мутации – синдром Дауна, нейрофиброматоз;
- вдыхание вредных веществ – бензола, алкила;
- прием медикаментов, угнетающих работу иммунной системы;
- ионизирующее облучение.
В группе риска по заболеванию находятся онкобольные и лица мужского пола, старше 60 лет.
Классификации и виды
Существует несколько классификаций заболевания согласно ВОЗ:
Рефракторная анемия:
В крови обнаруживается дефицит эритроцитов, при этом количество лейкоцитов и тромбоцитов остается в норме.
Рефракторная анемия с незрелыми ядросодержащими клетками костного мозга (с кольцевыми сидеробластами):
В анализах обнаруживается недостаток эритроцитов, с повышенным содержанием железа в клетках. Количество тромбоцитов и лейкоцитов в норме.
Рефракторная анемия с избыточным количеством бластов, находящихся на этапе трансформации:
Низкое количество эритроцитов к крови. Отмечается отклонение числа тромбоцитов и лейкоцитов от нормальных значений. В косном мозге наблюдается от 5-19% бластов.
Рефракторная цитопения с мультилинейной дисплазией:
Происходит снижение минимум двух ростков крови. В костном мозге выявляется до 5% бластов, в периферической крови – до 1%. Заболевание способно трансформироваться в лейкемию.
МДС с изолированным хромосомным отклонением:
В крови пациента недостаточно эритроцитов. В костном мозге определяется до 5% бластов, в анализах крови – до 1%. В хромосомах обнаруживаются специфические изменения.
Неклассифицируемый МДС:
В крови занижено количество тромбоцитов, эритроцитов или лейкоцитов. Количество бластов в крови и костном мозге в пределах нормы.
Первые признаки
Первые симптомы патологии – общая слабость и одышка. Иногда симптомы МДС никак не проявляют себя на начальных стадиях. Синдром выявляется случайно во время исследования крови в лаборатории. Первые признаки заболевания схожи по симптомам с патологиями печени и аутоиммунными нарушениями. К ним относится:
- бледность кожных покровов;
- возникновение синяков даже при незначительных травмах;
- образование точечных подкожных кровоизлияний;
- частые ОРВИ.
Зрелые симптомы
По мере прогрессирования болезни проявляются и другие признаки:
- быстрая утомляемость даже при легких физических нагрузках;
- ухудшение аппетита;
- потеря веса;
- подъем температуры до 40 градусов;
- трудности с дыханием при отсутствии астматических проявлений;
- обширные подкожные кровоподтеки;
- снижение гемоглобина в крови;
- болевые ощущения в костях и суставах;
- гепатомегалия.
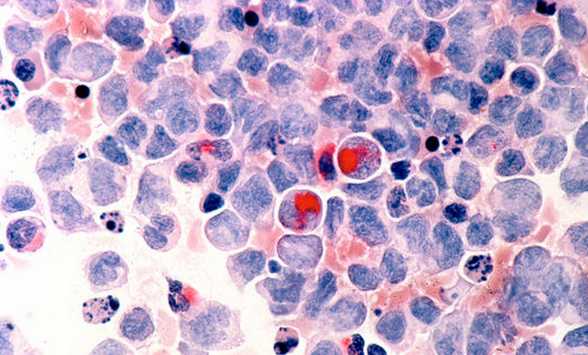
Что входит в диагностику?
Основные исследования для подтверждения заболевания – анализ крови на биохимию, цитогенетический и иммуногистохимический анализы клеток костного мозга. Кроме этого, при подозрении на МДС пациентам назначают следующие виды обследований:
Инструментальные методы диагностики позволяют оценить состояние других органов и систем. Перед постановкой диагноза в обязательном порядке проводится дифференциация, для того, чтобы исключить:
- острый лейкоз;
- злокачественные лимфомы;
- отравление организма ядовитыми веществами;
- миелодепрессивный синдром;
- проблемы с белковым обменом;
- патологии печени.
Как лечат сегодня?
Самый эффективный метод в борьбе с патологией – трансплантация костного мозга. Способ терапии имеет множество недостатков: высокую стоимость, вероятность отторжения трансплантата, необходимость дополнительной подготовки пациента к операции. Для осуществления процедуры трудно найти подходящего донора.
Перед трансплантацией стволовых клеток пациент должен пройти курс химиотерапии, который не всегда дает положительные результаты. Среди побочных эффектов процедуры отмечают: повреждение внутренних органов (особенно печени), выпадение волос. Для проведения химиотерапии чаще всего используется “Цитарабин” или “Децитабин”. Лекарства назначаются в высокой или низкой дозировке. Все зависит от возраста пациента, степени тяжести заболевания и общего состояния здоровья. Пожилые люди тяжело переносят данный вид лечения, поэтому химиотерапию им назначают только в крайних случаях.
Современный метод борьбы с МДС – трансплантация стволовых клеток. Побочные эффекты от процедуры такие же, как и при пересадке костного мозга. Данный способ позволяет предотвратить развитие острой лейкемии.
Немалую роль в борьбе с МДС играет переливание крови или сопроводительная терапия. Благодаря устраняется симптоматическая картина заболевания. Кровь вводится в организм больного в виде тромбоконцентрата или эритроцитарной массы. Сопроводительная терапия проводится в течение короткого промежутка времени, поскольку избыток железа в организме может дать побочные эффекты. Переливание крови нередко совмещают с медикаментозным лечением. К препаратам, стимулирующим выработку клеток крови, относят:
- “Нейпоген”;
- “Лейкин”;
- “Эритропоэтин”.
Также, при МДС назначаются лекарства для коррекции иммунитета, например, “Леналидомид”. С целью предупреждения лейкемии пациенту внутривенно вводят “Азацитидин”.
Прогноз и сколько живут такие пациенты?
Прогноз жизни при МДС, составленный ВОЗ, основывается на трех основных факторах:
- Кариотипе человека (по критерию ставится от 0-2 баллов).
- Разновидности патологии (по критерию ставится от 0-3 баллов).
- Необходимости в гемотрансфузии (от 0 -1 балла).
Баллы по трем критериям суммируются, и на основе показателя рассчитывается прогноз жизни больного МДС.
| Количество баллов | Прогноз выживаемости в месяцах |
| 0 | 136 |
| 1 | 63 |
| 2 | 44 |
| 3-4 | 19 |
| 5-6 | 8 |
Этот прогноз является приблизительным. Выживаемость пациента также во многом зависит от состояния здоровья и морального настроя.
Пациенты, которым был поставлен диагноз миелодиспластический синдром, задаются вопросом – может ли заболевание перерасти в рак. Однозначно ответить на этот вопрос невозможно, все зависит от типа проблемы и отношения человека к своему здоровью. Если пациент при возникновении первых признаков патологии обратился к специалисту, то с большей долей вероятности незрелые клетки не превратятся в злокачественные.