Миопатия дюшенна что это такое
симптомы, механизм развития и прогноз
Мышечная дистрофия, или миопатия Дюшенна, — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
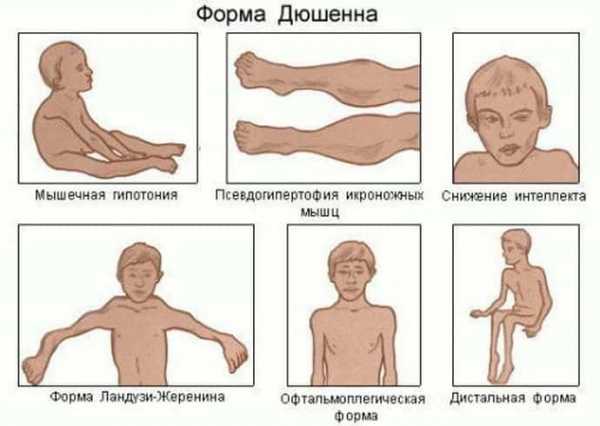
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.
Также миопатия Дюшенна формируется при заболеваниях соединительной ткани, не связанных напрямую с генетическими отклонениями.
Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
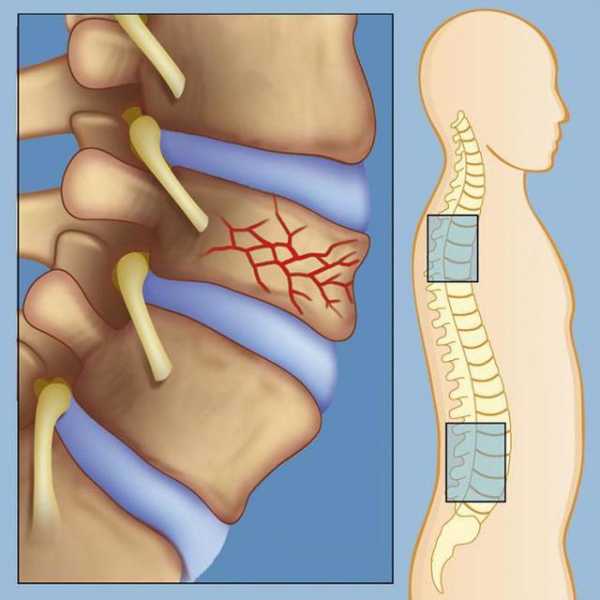
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.
Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:
- у ребенка возникает сильная неустойчивость, наблюдается неловкость в движении, он часто падает и очень медлителен;
- миопатия Дюшенна сопровождается тем, что во время ходьбы ребенок вихляет, постоянно спотыкается, в результате чего малыш боится вставать на ноги, возникает выраженная двигательная пассивность;
- со временем при мышечной дистрофии Дюшенна становится видна «утиная» походка с выпяченной вперед грудью и отведенными назад лопатками;
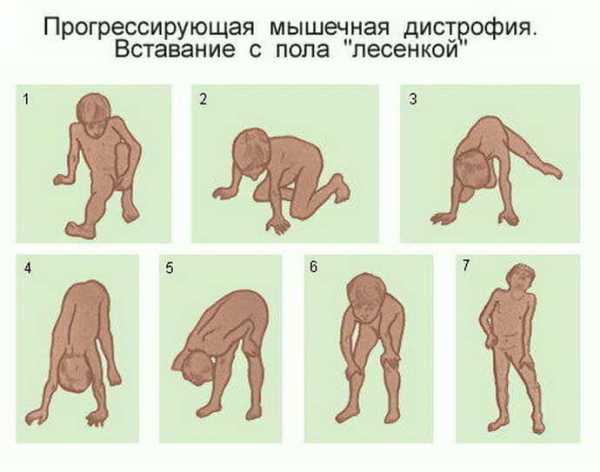
- если ребенок сидит или лежит, то принять стоячее положение при мышечной дистрофии Дюшенна становится сложно;
- при попытках принять стоячее положение, малыш словно встает на лесенку, поднимается вверх задом;
- возникает гипертрофия мышц, они заполняются жировой тканью;
- также миодистрофия Дюшенна захватывает работу сердца, в результате чего развиваются патологии и недостаточность;
- мышечная дистрофия Дюшенна часто сопровождается и другим признаком – появляются нарушения в биоптатах скелета;
- постепенно изменяется положение крупных суставов, начинается деформация стоп;
- миопатия Дюшенна в 100% случаев приводит к полной инвалидности пациента, ему требуется кресло;
- в 15 лет при мышечной дистрофии Дюшенна наступает глубокая инвалидизация, опасная остановкой сердца и хронические или постоянно рецидивирующие нарушения в легких.
На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.
Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.
Дополнительно нужно пройти ЭМГ, генодиагностику, а также биопсию мышц. Именно последний анализ позволяет установить болезнь с достаточно высокой точностью. Электромиография не уступает в эффективности с точки зрения постановки диагноза «мышечная дистрофия Дюшенна».
Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:
- Тактика при обнаружении болезни до 5 лет. Радикального лечения миодистрофии Дюшенна не требуется. Нужна генетическая консультация и постоянна поддержка родителей больного ребенка.
- Лечение миопатии Дюшенна до 8 лет. В этом случае нужна поддержка мышечных тканей. Врачи назначают глюкокортикостероиды для замедления прогресса болезни: «Преднизолон» или «Дефлазакорт».
- Терапия от 8 до 20 лет. В этом случае значительно ослабляются мышцы, мышечная дистрофия Дюшенна приводит ребенка к инвалидному креслу.
- Терапия от 20 лет. В этом случае препараты частично перестают действовать, прогрессируют заболевания дыхательных путей.
Миопатия Дюшенна требует постоянного приема некоторых групп витаминов (B, E), а также кальция, гормонов анаболиков, калия и некоторые виды аминокислот. Обязательно при мышечной дистрофии Дюшенна назначают инъекции АТФ, «Ретаболила», глютаминовой кислоты.
Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.
ЛФК проходят небольшими курсами с обязательным участием терапевта. Также врачи рекомендуют делать массаж. Для электрофореза при миопатии Дюшеннанеобходимо использовать такие вещества, как липаза, хлорид кальция, «Прозерин».
В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.

Мышечная дистрофия Дюшенна – тяжелое генетическое расстройство, терапия которого не способна оградить человека от одного исхода – смерти. В некоторых случаях пациентам удается прожить больше 20 лет после постановки диагноза. В других случаях младенцы умирают в течение первого года жизни.
Миодистрофия Дюшенна — Википедия
Материал из Википедии — свободной энциклопедии
Миодистрофия Дюшенна (другие названия: миопатия Дюшенна, миодистрофия Дюшенна-Беккера) — генетическая болезнь. Названа в честь французского врача-невролога Гийома Бенжамена Армана Дюшенна.
Болеют мальчики и очень редко девочки. Болезнь вызывается делециями или дупликациями одного или нескольких экзонов, либо точечными мутациями в гене дистрофина. Основное проявление — слабость мышц, затруднения при движениях с детского возраста, которые прогрессируют с течением времени. Признаками этого заболевания являются специфическая походка и осанка страдающих им мальчиков, позднее начало ходьбы, ухудшенная по сравнению со сверстниками речь, псевдогипертрофия икр[1]. Требуется лабораторная диагностика[2] — обычно речь идёт о биопсии мышечной ткани и генетическом исследовании. С 8-10 лет больным необходимы костыли, с 12 большинство из них прикованы к инвалидным коляскам, с 16-18 испытывают дыхательные нарушения. Также характерны поражения сердца (развивается кардиомиопатия[3]) и снижение интеллекта. Механизм развития последнего доподлинно не установлен. Смерть обычно наступает на втором-третьем десятилетии жизни. Средняя её продолжительность составляет 25 лет, однако есть люди, которые живут дольше[4].
Радикального лечения миодистрофии Дюшенна пока не предложено.
Существует только симптоматическое лечение. Стероиды способны замедлить развитие болезни[4]. Рекомендована лечебная физкультура. У больных повышен риск переломов костей. Требуется особая осторожность при назначении анестезии (наркоза) при хирургических вмешательствах.
В сентябре 2016 года FDA одобрило первый специализированный препарат — «Эксондис 51» (Exondys 51, этеплирсен), предназначенный для терапии мышечной дистрофии Дюшенна у пациентов с подтвержденной мутацией гена дистрофина, подходящей для корректировки посредством пропуска экзона 51. Таких больных насчитывается приблизительно 13% от общей популяции. Этеплирсен (eteplirsen), разработанный «Сарепта терапьютикс» (Sarepta Therapeutics), представляет собой антисмысловой олигонуклеотид, который связывается с экзоном 51 пре-мРНК дистрофина, чтобы исключать этот экзон в ходе мРНК-процессинга, тем самым реализуя альтернативный сплайсинг с последующим синтезом внутренне усеченного, но функционального дистрофина. Впрочем, терапевтическая эффективность этеплирсена в виде улучшения моторных функций по-прежнему остается недоказанной[5].
развитие и причины, проявления, лечение
Миопатия Дюшенна — наследственная патология, характеризующаяся прогрессирующим нарушением клеточного метаболизма в мышечной ткани, которое приводит к ее структурным изменениям и разрушению. Заболевание развивается только у мальчиков. Женщины – носители мутантного гена. Симптоматика болезни нарастает постепенно и начинает проявляться с самого детства. Родители замечают, что ребенок плохо стоит, с трудом карабкается на лесенку, часто падает во время ходьбы.
Миопатия была впервые описана в середине 19 века неврологом из Франции Г. Дюшенном. Он лечил больных с признаками мышечной патологии, анализировал наблюдения и представил свои умозаключения научному сообществу. Врач доказал генетическую природу заболевания.
В настоящее время существуют различные виды мышечных дистрофий. Миопатия Дюшенна считается наиболее распространенной среди них. Причиной патологии являются структурные перестройки хромосом: делеции или дупликации в гене белка дистрофина, который поддерживает целостность мембран мышечных волокон. Гипотрофический процесс носит восходящий характер: разрушаются мышцы ног, таза, спины, живота, плеча. В последнюю очередь возникает слабость рук. Но их пациенты еще долго могут использовать. Когда наступает паралич дыхательной мускулатуры и мышц головы, человек погибает.
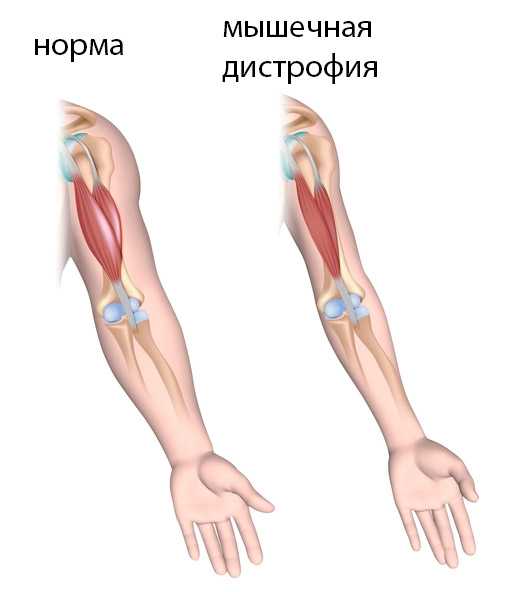
Мышечная слабость прогрессирует постепенно. Симптомами заболевания являются: специфическая походка, неправильная осанка, позднее физическое и психическое развитие, утрата приобретенных двигательных навыков. У детей возникают проблемы с ходьбой и бегом. С возрастом признаки патологии становятся более выраженными и разнообразными. Симптомы усугубляются под воздействием негативных факторов – психофизического перенапряжения, острых инфекций, отравления. Они резко снижают качество жизни лиц с миопатией. У больных отмечается гипо- и арефлексия, искривление позвоночника, килевидная или седловидная грудная клетка, контрактуры суставов и ретракции сухожилий. Часто к поражению опорно-двигательного аппарата присоединяются проблемы с сердцем в виде кардиомиопатии и изменения интеллекта в виде олигофрении и дебильности.
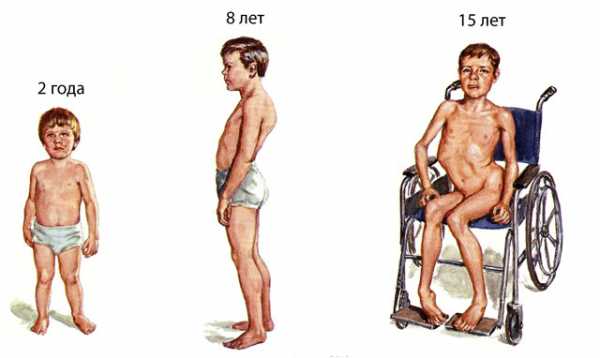
Диагностика миодистрофии Дюшенна заключается в проведении медико-генетического исследования, которое позволяет выявить мутантный ген. Больным проводят биопсию мышечной ткани для определения в ней уровня белка дистрофина. Данное наследственное заболевание неизлечимо. Побороть недуг невозможно. Специалисты проводят симптоматическую и паллиативную терапию, облегчающую и продлевающую жизнь больным. Медикаментозное лечение глюкокортикостероидами, ЛФК, массаж и физиопроцедуры в комплексе дают относительно неплохие результаты.
Заболевание отличается тяжелым, злокачественным течением и неблагоприятным прогнозом. Больные рано перестают самостоятельно двигаться, с 8-10 лет используют костыли, а с 12 – инвалидные коляски. Шестнадцатилетние подростки испытывают нарушения со стороны органов дыхания и сердца, у некоторых снижается интеллект. Нарастающая мышечная слабость приводит к полной обездвиженности больного. Замедлить разрушение мышц невозможно, что связано с врожденным характером патологических изменений. Лица с миодистрофией умирают в возрасте 25-30 лет от инфекционной пневмонии или острой коронарной недостаточности.
Этиология
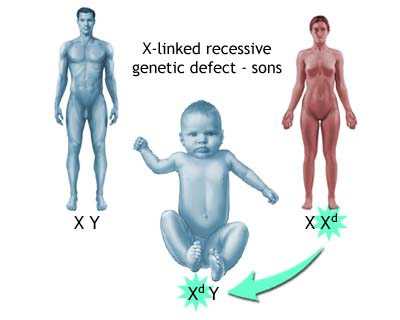 Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Мутантный ген, обуславливающий развитие миопатии, получил название ген Дюшенна. Он отвечает за синтез белка дистрофина, который обеспечивает целостность мышечных волокон во время сокращения. Белок составляет основу миофибрилл, поддерживает их клеточный скелет, позволяет им активно и многократно сокращаться и расслабляться. У лиц с миопатией он отсутствует или вырабатывается в очень незначительном количестве. Мышечные волокна постепенно разрушаются и замещаются фиброзной тканью или жиром. В результате серьезно страдает функция движения у больных.
Симптоматика
Новорожденные дети не имеют каких-либо явных отклонений в здоровье и строении. Первые клинические признаки появляются у малышей 1,5-2 лет. Болезнь прогрессирует, а симптоматика нарастает.
проявления миопатии Дюшенна
- Родители замечают, что их ребенок двигается неловко и неустойчиво, постоянно спотыкается и падает при ходьбе, не может прыгнуть, подняться по ступенькам, встать из лежачего или сидячего положения. Больные дети очень медлительны и неуклюжи во время двигательной активности. Это связано с поражением мышц таза и ягодиц, которые становятся слабыми первыми.
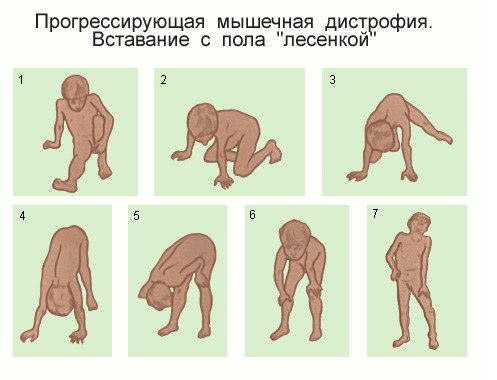
- Появляется характерный симптом Говерса: больные, поднимаясь с пола, помогают себе руками – они опираются на свои колени и бедра.
- Еще один патогномоничный признак миопатии Дюшена – ложная гипертрофия мышц голени, бедер, ягодиц: они кажутся большими, упругими, твердыми. На самом деле мышечная ткань разрушилась, а на ее месте разрослась фиброзная и жировая ткань.
- Больные дети испытывают страх перед ходьбой и с каждым днем становятся все более пассивными. Они с трудом стоят без помощи окружающих, быстро утомляются и отличаются низкой выносливостью. Мышечная слабость в ногах становится причиной формирования «утиной» походки — больные как бы переваливаются с ноги на ногу. При прогрессировании миопатии патологический процесс охватывает весь торс, в том числе и верхние конечности.
- У больных возникает поражение миокарда, протекающее в форме кардиомиопатии, причиной которой является также дефицит дистрофина. У детей возникают болезненные ощущения в груди, дыхание становится глубоким, неритмичным и частым. Даже на ранней стадии патологии появляются изменения на электрокардиограмме, характерные для коронарной недостаточности. Сердечно-сосудистые расстройства проявляются аритмией, лабильностью артериального давления, приглушенностью сердечных тонов.
- У части пациентов нарушается психоэмоциональное развитие, проявляющееся отставанием в обучении от ровесников, речевым расстройством в виде дислексии, плохой памятью. У 30% больных возникает олигофрения, аутизм. Интеллектуальные возможности у таких детей постепенно снижаются, порой достигая легкой степени идиотии. Невозможность посещать детские сады, школы и прочие общественные заведения еще больше усугубляет имеющиеся когнитивные расстройства.
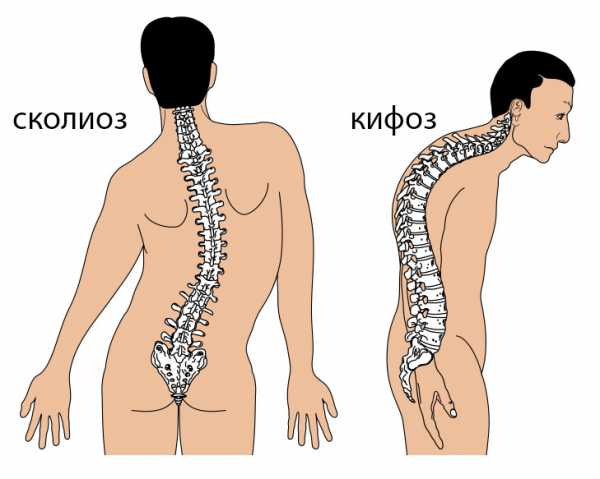 При прогрессировании патологии развиваются контрактуры крупных суставов, деформируется стопа, искривляется позвоночник по типу кифоза или лордоза, резко слабеют, а затем и полностью исчезают сухожильные коленные рефлексы.
При прогрессировании патологии развиваются контрактуры крупных суставов, деформируется стопа, искривляется позвоночник по типу кифоза или лордоза, резко слабеют, а затем и полностью исчезают сухожильные коленные рефлексы.- Миодистрофия Дюшенна всегда сопровождается повреждением грудины, которая приобретает неправильную форму и сильно изменяет тело человека. Из-за тяжелых атрофических процессов в мышцах талия становится «осиной», а лопатки «крыловидными».
- У 50% больных возникают эндокринные расстройства, проявляющиеся ожирением, гипоплазией и гипофункцией половых органов, низкорослостью.
Больные дети рано становятся инвалидами, прикованными к креслу. К 10-12 годам они полностью утрачивают способность к самостоятельному передвижению, а к 15 — к совершению любых действий. В течение пяти лет после этого атрофируются дыхательные мышцы.
Погибают больные обычно в возрасте 25 лет от неуклонно нарастающей слабости скелетных мышц, сформировавшейся стойкой дисфункции органов дыхания и сердечно-сосудистой системы.
Осложнения
Мышечная дистрофия Дюшенна сокращает жизненный путь человека. Это основное и самое страшное последствие болезни.
Осложнения со стороны опорно-двигательного аппарата:
- Остеопороз — уменьшение плотности костной ткани,
- Патология суставов — снижение их подвижности из-за сильной мышечной слабости,
- Сколиоз, кифоз, лордоз — различные формы искривления позвоночника.
Поражение органов пищеварения:
- Запоры — результат гиподинамии,
- Потеря веса из-за разрушения мышц,
- Нарушение процесса жевания и глотания требует питания больного через зонд.
Дыхательные расстройства:
- Поверхностное дыхание,
- Слабый кашлевой рефлекс,
- Частые ОРВИ,
- Недостаток кислорода в крови – головные боли по утрам, пробуждения по ночам, слабость, раздражительность, насыщенные сновидения.
У лиц с миопатией Дюшена развивается кардиомиопатия — слабость миокарда, проявляющаяся повышенной утомляемостью, одышкой, отеками ног, перебоями в работе сердца.
Своевременная диагностика и эффективная терапия могут отсрочить развитие осложнений или совсем предотвратить их появление.
Диагностические мероприятия
Диагностика синдрома Дюшенна не вызывает трудностей у специалистов, поскольку имеет весьма специфические симптомы.
Врачи традиционно начинают с опроса больного или его родителей и сбора анамнестических данных. Особое внимание они уделяют:
- Времени появления первых симптомов,
- Локализации первичной мышечной слабости,
- Общему самочувствию пациента,
- Наличию подобных расстройств у родных и близких.
Во время неврологического обследования выявляется:
- Слабость определенной группы мышц и определяется ее степень,
- Изменение мышечного тонуса,
- Атрофические процессы,
- Гипо- и арефлексия,
- Деформация стопы, груди, позвоночного столба.
Врачи наблюдают за больным ребенком, обращая внимание на то, как он ходит, бегает и встает с пола. Изменение походки — важный диагностический признак миопатии.
После проведения первичных диагностических процедур врачи могут заподозрить наличие у больного патологии и поставить предварительный диагноз. Чтобы его подтвердить или опровергнуть, пациента направляют на лабораторно-инструментальное обследование.
В лаборатории проводят следующие исследования:
- Анализ крови на гормональный статус.
- Биохимический анализ крови на активность креатинкиназы – фермента, уровень которого у больных детей очень высок. Если КФК в норме, миопатию Дюшена исключают.
- Иммуногистохимическое исследование – микроскопия биоптата мышечной ткани, взятого от больного, с целью определения белка дистрофина. У лиц с миопатией он отсутствует.
- ДНК-тест – генетическое исследование крови больного, позволяющее определить мутантный ген и точно диагностировать патологию.
Дополнительные диагностические методики:
- Электрокардиография — выявление признаков поражения миокарда.
- Электромиография — определение фибрилляции, свидетельствующей о некрозе мышечных волокон. Эта методика оценивает состояние скелетной мускулатуры и подтверждает, что в основе патологии лежит именно поражение мышц, а не нарушение передачи нервных импульсов.
- Дыхательные пробы, рентгенография позвоночника и органов грудной клетки, УЗИ сердца — методы, не оказывающие существенного влияния на процесс постановки диагноза, но позволяющие выявить имеющиеся отклонения в структуре и функционировании органов дыхания и сердца.
Лечебный процесс
Болезнь Дюшенна, как и любая другая наследственная патология, неизлечима. Врачи проводят симптоматическую терапию, устраняющую неприятные проявления, предупреждающую развитие осложнений и продлевающую жизнь больных.
Комплексное поддерживающее лечение болезни:
- Медикаментозная терапия замедляет, но не останавливает прогрессирование миопатии. Больным назначают глюкокортикостероидные препараты «Преднизолон», «Бетаметазон», сердечные средства «Ритмонорм», «Анаприлин», «Периндоприл», метаболические препараты «Милдронат», «Рибоксин», «Циннаризин», поливитамины «Нейромультивит», «Доппельгерц».
- Физиотерапия заключается в проведении пассивного растяжения пораженных мышц, ЛФК, массажа, электрофореза, гидротерапии, индуктотермии, лазеротерапии, ультрафонофореза.
- Диетотерапия – полноценное питание с достаточным количеством растительных жиров и животных белков. Больным рекомендуют исключить из рациона крепкий чай и кофе, спиртное, пряности, сдобу, шоколад, фаст-фуд. Полезными являются блюда из овощей и фруктов, молочнокислые продукты, крупяные изделия, яйца.
- Чтобы поддержать функционирование органов дыхания на оптимальном уровне, больным необходима искусственная вентиляция легких, насыщающая кровь кислородом. Для этого в домашних условиях используют различные портативные аппараты, а в стационаре тяжелобольным пациентам интубируют трахею или выполняют трахеостомию.
- Ортопедическая помощь необходима всем больным с миопатией Дюшенна. Причем, чем старше становится ребенок, тем ярче выражены признаки патологии, а значит и потребность в ортопедических приспособлениях гораздо больше. Вертикализаторы, инвалидные кресла, специальные шины и корсеты существенно облегчают жизнь больным лицам.
- В отдельных случаях пациентам требуется психотерапевтическая поддержка. Это связано с развитием у детей депрессии и появлением суицидальных наклонностей. Не каждый ребенок в подростковом возрасте может смириться со своей беспомощностью и жизнью в инвалидном кресле.
- Применение стволовых клеток и генная терапия — развивающиеся направления в лечении наследственных заболеваний. Путем введения в организм здоровых генов или замены обычных клеток стволовыми можно улучшить работу организма на клеточном уровне и нормализовать состояние больного. С помощью стволовых клеток восстанавливают пораженные мышечные волокна. Больным подсаживают миогенные клетки, вырабатывающие нормальный дистрофин в достаточном количестве. Любая научная разработка, доказанная опытным путем, дает надежду больным на выздоровление.
Профилактика и прогноз
Супругам, в роду которых имелись случаи наследственных заболеваний, перед планированием беременности необходимо посетить врача-генетика. Профилактика патологии также заключается в проведении пренатальной диагностики. Выявив миопатию на ранних сроках, можно прервать беременность.
Миопатия Дюшена — наследственная патология, отличающаяся тяжелым течением и быстрым прогрессированием. Это современная медицинская проблема, характеризующаяся разрушением мышечной ткани и быстрым развитием мышечной слабости. Все без исключения больные погибают в раннем возрасте из-за развития не совместимых с жизнью осложнений. Только адекватная и комплексная терапия, четкое соблюдение рекомендаций врача, тщательный уход и забота родителей могут замедлить ход болезни.
Больные быстро становятся инвалидами и погибают в совсем юные годы. Самое страшное, что детям с миопатией не в силах помочь даже квалифицированные врачи, современные медицинские технологии и терапевтические методики 21 века. Болезнь до сих пор остается неизлечимой, забирая молодые жизни. Современные ученые-медики всего мира трудятся над созданием радикального способа борьбы с миопатией Дюшенна.
Видео: ребенок с синдромом Дюшенна
Видео: о перспективном методе терапии МД
причины, симптомы, лечение, как наследуется дистрофия, ее диагностика, фото
Мышечные дистрофии – это группа заболеваний не воспалительного характера, характеризующиеся прогрессивным течением без патологических изменений центральной и периферической нервной системы.
Что это такое?
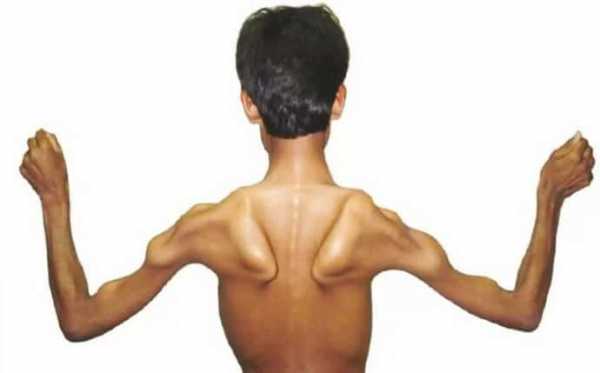
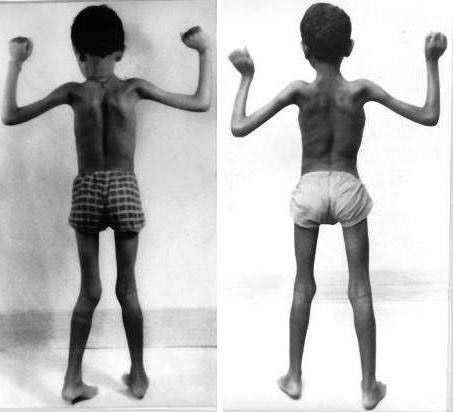
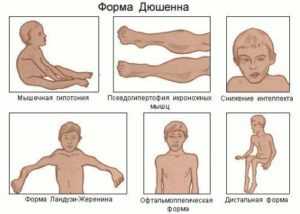 Фото 1. Признаки миопатии Дюшенна
Фото 1. Признаки миопатии ДюшеннаПервые публикации о миопатиях датируются 1830 годом. В 1852 году Мерион сообщил о случае в семье с четырьмя сыновьями, у которых отмечались признаки двигательных расстройств, не связанных с поражением нервной системы. Он предположил, что заболевание наследуется от матери к ребенку.
Под миопатией или миодистрофией Дюшенна (также ее называют дистрофией или миопатией Дюшена-Беккера) понимают заболевание генетической природы, связанное с мутацией гена, кодирующего синтез белка дистрофина.
Дистрофин входит в состав мембран мышечных клеток, без него мышцы крайне уязвимы и подвергаются некрозу и дегенерации. Экспрессия, то есть реализация функции, происходит в гладкой и скелетной мышечной ткани, а также в миокарде.
Миопатия Дюшенна поражает одного из 3500 новорожденных детей. Несмотря на тот факт, что заболевание названо именем Дюшенна, одним из первых врачей, описавших данную патологию, был Говерс.
Гийом Дюшенн был французским неврологом, который использовал электростимуляцию в лечении неврологических расстройств. В 1868 году Дюшенн описал 13 пациентов с заболеванием, которое сопровождалось прогрессирующей мышечной слабостью, и назвал ее паралитической псевдогипертрофической мышечной дистрофией. Также он определил диагностические критерии миодистрофии, которые используются и сегодня.
Этиология, кто наследует и почему
По международной классификации болезней МКБ-10 мышечная дистрофия Дюшенна-Беккера имеет код G71.
Одна треть случаев заболевания связана со спонтанно возникшей мутацией, в то время как на остальные случаи патологии приходится наследование Х-хромосомы, несущей патологический ген.
Случаи гонадного мозаицизма связаны с 20% случаев миодистрофии Дюшенна. Средняя продолжительность жизни менее 20 лет. Повышение уровня фермента креатинфосфокиназы (КФК) обнаруживают у 2/3 женщин – носителей патологического гена. Большинство из них не имеют никаких проявлений заболевания. Миопатия почти всегда поражает мальчиков, поскольку заболевание сцеплено с Х-хромосомой, тип наследования – рецессивный. Локализация гена, кодирующего синтез дистрофина, находится в локусе Xq21. Синтез белка кодируется одним из самых больших известных генов. Он занимает около 2% ДНК Х-хромосомы.
Симптомы мышечной дистрофии
Первые симптомы появляются в возрасте от трех до семи лет. Обычно родители замечают раскачивающуюся походку и гиперлордоз. Существует несколько основных критериев, которые позволяют предположить миопатию Дюшенна. К ним относятся:
- Мышечная слабость, которая появляется неожиданно и начинается с нижних конечностей.
- Гиперлордоз – выраженный изгиб позвоночника кпереди, что особенно проявляется при ходьбе.
- Гипертрофия ослабленных мышц.
- Слабый ответ мышц на электрическую стимуляцию на более поздних стадиях заболевания.
С течением времени все указанные симптомы прогрессируют. Несмотря на поражение мышечных структур, нарушений функции мочевого пузыря и кишечника не отмечается. К двенадцати годам жизни большинство пациентов не могут ходить самостоятельно, и находится в инвалидном кресле. Миопатия Беккера очень сходна с дистрофией Дюшенна, так как поражается один и тот же локус Х-хромосомы, в результате чего страдает синтез дистрофина. Отличием миопатии Беккера является начало заболевания, как правило, после трех лет жизни или даже в подростковом возрасте.
Миодистрофия Дюшенна – патология, которая затрагивает не только скелетную мускулатуру. Дистрофин также содержится в миокарде, тканях мозга и гладкомышечной мускулатуре. Поздняя стадия заболевания ассоциируется с тяжелой сердечной недостаточностью и дыхательными нарушениями – главными причинами смерти пациентов.
Формы миопатии
В течении заболевания различают пять стадий. Первая стадия (доклинических проявлений) не характеризуется широким спектром симптомов, обычно у пациентов отмечается лишь повышение уровня КФК в сыворотке крови.
Вторая стадия (ранних признаков) включает следующие симптомы:
- Раскачивающаяся походка – впервые появляется от 2-х до 6-ти лет и часто является первым симптомом, который замечают родители.
- Прогрессирующая слабость мускулатуры нижних конечностей с дальнейшим присоединением слабости мышц шеи, плечевого пояса и рук.
- Наличие симптома Говерса – при попытке встать с пола, пациент упирается на колени и руки (рисунок 1). Появление симптома связано с выраженной слабостью мышц спины и конечностей.
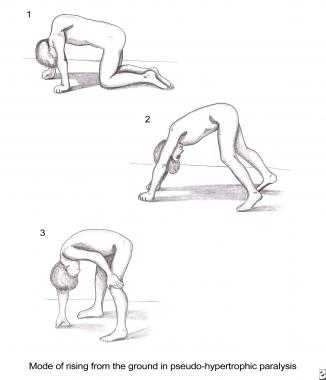 Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки.
Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки.Третья стадия (прогрессирующих симптомов) характеризуется появлением значительных сложностей во время ходьбы и развивается, когда возраст ребенка составляет около восьми лет. Пациенту становится сложно подниматься по ступеням, появляется одышка, сложно встать с пола. По утрам могут беспокоить головные боли, в ночное время наблюдается затрудненное дыхание. Четвертая и пятая стадии являются наиболее тяжелыми, так как пациент теряет возможность не только самостоятельно передвигаться, но и испытывает трудности с дыханием. При четвертой стадии пациент еще может удерживать осанку, однако делать это становится все труднее, развивается выраженный сколиоз.
Пятая стадия – терминальная. Пациент не может ходить и, как правило, находится в инвалидном кресле. Чаще всего, смерть наступает от дыхательной и сердечно-сосудистой недостаточности в возрасте до 20-ти лет.
Методы диагностики
Ребенок с миопатией Дюшенна-Беккера до 2-3-х лет жизни может ничем не отличаться от других детей. Тем не менее, существует ряд признаков, на которые стоит обратить внимание.
Важно! Во время проведения лабораторных исследований может быть повышен уровень печеночных ферментов: аланин- и аспартатаминотрансферазы, креатинкиназы и гамма-глутаматтрансферазы.
В ряде исследований миопатий было отмечено, что задержка речи и моторных навыков отмечалась чаще у детей с дефектом дистрофина. Уровень IQ может быть меньше на одно стандартное отклонение по сравнению со среднестатистическим значением в популяции.
У 30% детей с мутациями гена, кодирующего синтез дистрофина, отмечались трудности с обучением и приобретением новых навыков, обсессивно-компульсивные расстройства, синдром дефицита внимания, задержка умственного развития. Детям с дистрофиями Дюшенна и Беккера сложнее даются вербальные навыки.
Постановка диагноза «Миопатия Дюшенна» включает:
- Исследование уровня КФК, которая превышает референсные значения в 50-100 раз.
- Молекулярную диагностику – исследование гена, локализованного в локусе Xq21 и ответственного за синтез дистрофина, позволяет подтвердить или опровергнуть диагноз.
Если у пациента наблюдается сочетание высоких показателей КФК и мышечная слабость, это с высокой вероятностью позволяет заподозрить миопатию Дюшенна-Беккера.
Из инструментальных методов используют:
- обзорную рентгенографию грудной клетки. Исследование позволяет сделать заключение, насколько выражен сколиоз;
- электромиографию: метод применяют с целью дифференциальной диагностики со спинальной мышечной атрофией;
- электрокардиография – с целью выявления синусовых аритмий;
- ультразвуковое исследование сердца – нередко определяет малые размеры желудочков сердца и более длинную диастолу;
- холтеровское мониторирование определяет наличие пароксизмальных аритмий.
Если молекулярная диагностика не выявила мутации гена дистрофина, рекомендуется провести биопсию мышечной ткани. Характерные гистологические изменения приведены ниже:
- мышечные волокна с выраженным дегенеративным процессом и некрозом;
- пролиферация соединительной ткани;
- появление жировой ткани в значительном количестве.
Также проводится анализ белка дистрофина, выделенного из мышцы, с определением его молекулярной массы.
Лечение заболевания
На сегодняшний день лечения миодистрофии Дюшенна-Беккера не существует. Терапевтическая тактика основана на поддерживающем медикаментозном лечении (в основном, для сохранения сердечной функции), исследованиях в области генной инженерии и экспериментальном клеточном лечении.
При прогрессировании сколиоза и контрактур суставов возможно применение паллиативного хирургического вмешательства. По мере прогрессирования мышечной слабости, следует обеспечить пациенту максимальный комфорт.
Применение преднизолона активно обсуждается в профессиональных медицинских сообществах. Известно, что спустя один месяц от начала применения наблюдается незначительное улучшение общего состояния пациента и может сохраняться до трех лет без ухудшения. В то же время, при отказе от преднизолона начинается прогресс заболевания.
Последствия и осложнения
Главными осложнениями, с которыми сталкиваются пациенты, являются:
- Прогрессирующая мышечная слабость.
- Дилатационная кардиомиопатия.
- Дыхательные нарушения, связанные с дисфункцией диафрагмы.
- Контрактуры суставов.
- Сколиоз.
- Дисфагия.
- Запоры.
К осложнениям также относится остеопороз и высокий риск переломов. По некоторым данным возможно использование препаратов кальция и витамина Д для увеличения плотности костной ткани.
Прогноз на выздоровление и жизнь
К сожалению, прогноз неблагоприятный, так как смертность при данной патологии составляет 100%. Если в семейной истории встречается близкая родственница, которая является носителем патологического гена, у детей мужского пола выше риск развития кардиомиопатии с прогрессирующей сердечной недостаточностью в возрасте от 20 до 40 лет. Проводятся исследования, связанные с применением стволовых клеток для лечения миодистрофий.
Что нужно запомнить?
- Миопатия Дюшенна-Беккера – прогрессирующее заболевание, связанное с аномальным синтезом белка дистрофина, принимающего непосредственное участие в мышечных сокращениях. Синтез дефектного белка приводит к дегенеративным и некротическим процессам в мышцах.
- Ранними признаками болезни считаются появление мышечной слабости в нижних конечностях, раскачивающейся, нетипичной походки. К лабораторным признакам относится многократное повышение уровня КФК.
- Этиология заболевания – генетическая, связанная с мутацией гена в локусе Тип наследования-рецессивный, мутация может передаваться от матери, чья Х-хромосома несет патологический ген, а может возникать спонтанно.
- Существует пять стадий миопатии. Четвертая и пятая стадии являются терминальными.
- К ранним методам диагностики относится определение уровня КФК, который может многократно превышать норму и являться ранним маркером наследственной миодистрофии Дюшенна-Беккера задолго до появления мышечной слабости и других заметных симптомов. К более точным методам относится молекулярная диагностика с исследованием патологического гена.
- Специфическое лечение не разработано. Мероприятия по уходу за пациентом сводятся к поддержанию жизнеобеспечения.
- Основными осложнениями являются утрата способности ходить, тяжелые дыхательные расстройства и сердечно-сосудистая недостаточность.
- Прогноз неблагоприятный, летальность составляет 100%.
Литература
- Bushby K, Straub V. Nonmolecular treatment for muscular dystrophies. Curr Opin Neurol. 2005 Oct. 18(5):511-8.
- Moxley RT 3rd, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005 Jan 11. 64(1):13-20.
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991 Sep 20. 66(6):1121-31.
- Ozawa E, Noguchi S, Mizuno Y, et al. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998 Apr. 21(4):421-38.
- Darke J, Bushby K, Le Couteur A, McConachie H. Survey of behaviour problems in children with neuromuscular diseases. Eur J Paediatr Neurol. 2006 May. 10(3):129-34.
- Mendell JR, Shilling C, Leslie ND et al. Evidence based path to newborn screening for Duchenne Muscular Dystrophy. Ann Neurology. 2012. 71:304–313.
- Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007 Sep. 64(9):1236-41.
- Merlini L, Gennari M, Malaspina E et al. Early corticosteroid treatment in 4 duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012 Jun. 45(6):796-802.
- American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005 Dec. 116(6):1569-73.
- Colan SD. Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation. 2005 Nov 1. 112(18):2756-8.
Мышечная дистрофия (миопатия): симптомы и лечение, что такое мышечная дистрофия Дюшенна
Закрыть- Болезни
- Инфекционные и паразитарные болезни
- Новообразования
- Болезни крови и кроветворных органов
- Болезни эндокринной системы
- Психические расстройства
- Болезни нервной системы
- Болезни глаза
- Болезни уха
- Болезни системы кровообращения
- Болезни органов дыхания
- Болезни органов пищеварения
- Болезни кожи
- Болезни костно-мышечной системы
- Болезни мочеполовой системы
- Беременность и роды
- Болезни плода и новорожденного
- Врожденные аномалии (пороки развития)
- Травмы и отравления
- Симптомы
- Системы кровообращения и дыхания
- Система пищеварения и брюшная полость
- Кожа и подкожная клетчатка
- Нервная и костно-мышечная системы
- Мочевая система
- Восприятие и поведение
- Речь и голос
- Общие симптомы и признаки
- Отклонения от нормы
- Диеты
- Снижение веса
- Лечебные
- Быстрые
- Для красоты и здоровья
- Разгрузочные дни
- От профессионалов
- Монодиеты
- Звездные
- На кашах
- Овощные
- Детокс-диеты
- Фруктовые
- Модные
- Для мужчин
- Набор веса
- Вегетарианство
- Национальные
- Лекарства
- Антибиотики
- Антисептики
- Биологически активные добавки
- Витамины
- Гинекологические
- Гормональные
- Дерматологические
- Диабетические
- Для глаз
- Для крови
- Для нервной системы
- Для печени
- Для повышения потенции
- Для полости рта
- Для похудения
- Для суставов
- Для ушей
- Желудочно-кишечные
- Кардиологические
- Контрацептивы
- Мочегонные
- Обезболивающие
- От аллергии
- От кашля
- От насморка
- Повышение иммунитета
- Противовирусные
- Противогрибковые
- Противомикробные
- Противоопухолевые
- Противопаразитарные
- Противопростудные
- Сердечно-сосудистые
- Урологические
- Другие лекарства
- Врачи
- Клиники
- Справочник
- Аллергология
- Анализы и диагностика
- Беременность
- Витамины
- Вредные привычки
- Геронтология (Старение)
- Дерматология
- Дети
- Женское здоровье
- Инфекция
- Контрацепция
- Косметология
- Народная медицина
- Обзоры заболеваний
- Обзоры лекарств
- Ортопедия и травматология
- Питание
- Пластическая хирургия
- Процедуры и операции
- Психология
- Роды и послеродовый период
- Сексология
- Стоматология
- Травы и продукты
- Трихология
- Другие статьи
- Словарь терминов
- [А] Абазия .. Ацидоз
- [Б] Базофилы .. Богатая тромбоцитами плазма
- [В] Вазопрессин .. Выкидыш
- [Г] Галлюциногены .. Грязи лечебные
- [Д] Деацетилазы гистонов .. Дофамин
- [Ж] Железы .. Жиры
- [И] Иммунитет .. Искусственная кома
- [К] Каверна .. Кумарин
- [Л] Лапароскоп .. Лучевая терапия
- [М] Макрофаги .. Мутация
- [Н] Наркоз .. Нистагм
- [О] Онкоген .. Отек
- [П] Паллиативная помощь .. Пульс
- [Р] Реабилитация .. Родинка (невус)
- [С] Секретин .. Сыворотка крови
- [Т] Таламус .. Тучные клетки
- [У] Урсоловая кислот
Миопатия Дюшена: Диагноз. Лечение. Осложнения.
- О САЙТЕ
- НАГРАДЫ
- БАННЕРЫ
- КАРТА САЙТА
- ОБРАТНАЯ СВЯЗЬ
- О САЙТЕ
- НАГРАДЫ
- БАННЕРЫ
- КАРТА САЙТА
- ОБРАТНАЯ СВЯЗЬ
- ГЛАВНАЯ
- НОВОСТИ
- АКТУАЛЬНО
- ЗАКОН И ПРАВО
- ОНЛАЙН-ТРАНСЛЯЦИИ
- ВИДЕОГАЛЕРЕЯ
- ФОТОХРОНИКА
- Смерть пациента с миодистрофией Дюшенна в связи с приемом глюкокортикостероидов 7 часов ago
- Roche подала в Минздрав заявку на регистрацию препарата для лечения СМА. Он станет вторым в России 7 часов ago
- Ольга Германенко: “Не важно, какой препарат принимать при СМА, — важно начать прием как можно быстрее” 2 дня ago
- ИССЛЕДОВАНИЯ
- МНЕНИЕ СПЕЦИАЛИСТОВ
- МИОДИСТРОФИЯ ДЮШЕННА
- СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ
- ЭКСОНДИС 51 / EXONDYS 51
- АТАЛУРЕН / TRANSLARNA
- СПИНРАЗА / SPINRAZA (NUSINERSEN)
- EMFLAZA / ДЕФЛАЗАКОРТ
- CRISPR/Cas9
- СТВОЛОВЫЕ КЛЕТКИ
- ОБМЕН ОПЫТОМ
- ПЕРСОНАЛИИ
- ИНТЕРВЬЮ
- В кругосветное из Беларуси: человек с инвалидностью о инклюзии, путешествиях и супергероях 5 дней ago
- «Решила вернуться к прошлому»: как живёт певица Юлия Самойлова после Евровидения 6 дней ago
- «Иногда у ребенка нет и дня». В России больше двух тысяч больных СМА. Им нужна помощь государства 2 недели ago
- ИНВАИННОВАЦИИ
- НАУКА И ТЕХНОЛОГИИ
- ОБЗОРЫ ТЕХ.СРЕДСТВ
- «Мне писали: „Вы убиваете детей!“» Пары, сделавшие ЭКО, — об осуждении общества и своих детях 2 месяца ago
- Дмитрий Шевцов – о платной медицине, пенсионном возрасте, эвтаназии в Беларуси и работе депутатом 2 месяца ago
- Как ученый решил превратиться в киборга: союз машины и плоти 3 месяца ago
- БИБЛИОТЕКА
- МИОЛИТЕРАТУРА
- СЕМЬЯ И ДЕТИ
- КАРДИОЛОГИЯ
- ФИЗИОЛОГИЯ
- ПСИХОЛОГИЯ
- Побороть неизлечимое. Как поддерживают пациентов, страдающих СМА? - 3 месяца ago
что это, причины, симптомы, лечение
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна (МДД) — это генетическое заболевание, характеризующееся прогрессирующей мышечной дегенерацией и слабостью. Это один из девяти видов мышечной дистрофии.
МДД вызывается отсутствием дистрофина, белка, который помогает сохранить мышечные клетки в целости и сохранности. Симптом начинается в раннем детстве, обычно в возрасте от 3 до 5 лет.
Болезнь в основном поражает мальчиков, однако в редких случаях может поражать и девочек.
Мышечная слабость может начаться уже в возрасте 3 лет, сначала поражая мышцы бедер, тазовой области и плеч, а затем скелетные (произвольные) мышцы рук, ног и туловища. В раннем подростковом возрасте также поражаются сердце и дыхательные мышцы.
Причины мышечной дистрофии Дюшенна
До 1980-х годов было мало что известно о причинах какой-либо формы мышечной дистрофии. В 1986 году проводившиеся исследования, идентифицировали ген на Х-хромосоме у больных с МДД, который, будучи дефектным (мутировавшим), вызывает как мышечные дистрофии Дюшенна, так и Беккера (более слабый тип МДД).
Гены содержат коды или рецепты белков, которые являются важными биологическими компонентами во всех формах жизни. В 1987 году белок, связанный с этим геном, был идентифицирован и назван дистрофином.
МДД возникает из-за того, что мутированный ген не способен продуцировать практически любой функциональный дистрофин.
Недостаток дистрофина вызывает повреждение мышц и прогрессирующую слабость, начиная с раннего детства.
Белок дистрофина передает силу мышечного сокращения изнутри мышечной клетки наружу к клеточной мембране. Поскольку он соединяет центр мышечной клетки с периферией, белок дистрофина чрезвычайно длинный. Один конец специализирован для связывания с внутренней частью мышцы, а другой — для связывания с различными белками на клеточной мембране. Длинный средний участок, называемый стержневым доменом, занят серией повторяющихся звеньев, называемых повторениями спектрина.
Повторяющиеся единицы спектранов в середине белка играют важную роль в связывании двух концов, но исследования показали, что точное количество этих единиц не является критическим для функции белка в целом.
Многие случаи МДД вызваны мутациями в той части гена, которая кодирует эту среднюю часть. Производство всего белка прекращается, когда встречается мутация.
Отсутствие дистрофина приводит в движение каскад вредных воздействий. В мышцах начинает формироваться фиброзная ткань, а иммунная система организма усиливает воспаление. В дополнение к своей роли передачи силы дистрофин обеспечивает основу для удержания многочисленных молекул на месте вблизи клеточной мембраны. Потеря дистрофина вытесняет эти молекулы с последующим нарушением их функций.
Наследование

Мышечная дистрофия Дюшенна наследуется по X-сцепленной схеме, потому что ген, который может нести вызывающую МДД мутацию, находится в X-хромосоме. Каждый мальчик наследует Х-хромосому от своей матери и Y-хромосому от своего отца, что делает его мужчиной. Девочки получают две Х-хромосомы, по одной от каждого родителя.
Каждый сын, родившийся от женщины с мутацией дистрофина в одной из двух ее Х-хромосом, имеет 50-процентную вероятность наследования дефектного гена и наличия МДД. Каждая из ее дочерей имеет 50-процентный шанс унаследовать мутацию и быть носителем. Носители могут не иметь никаких симптомов заболевания, но могут иметь ребенка с мутацией или заболеванием. Носители МДД подвержены риску кардиомиопатии.
Несмотря на то, что МДД часто встречается в семье, у семьи без истории МДД может внезапно появиться сын с этой болезнью. Этому есть два возможных объяснения:
- Генетическая мутация, приводящая к МДД, могла существовать в семье женщины в течение нескольких поколений, и никто об этом не знал. Возможно, ни один ребенок мужского пола не рождался с этой болезнью, или, даже если бы мальчик более раннего поколения был больным, родственники, возможно, не знали, какое у него заболевание.
- Вторая возможное объяснение заключается в том, что у ребенка с МДД появилась новая генетическая мутация, возникшая в одной из яйцеклетки матери. Поскольку этой мутации нет в клетках крови матери, ее невозможно обнаружить с помощью стандартного генетического теста.
Если мать рожает ребенка с мышечной дистрофией Дюшенна, всегда существует вероятность того, что более чем в одной из ее яйцеклеток имеется мутация гена дистрофина, что подвергает ее риску передачи мутации другому ребенку выше среднего. И как только новая мутация будет передана сыну или дочери, он или она может передать ее следующему поколению.
Мужчина с МДД не может передать дефектный ген своим сыновьям, потому что передает сыну Y-хромосому, а не X. Но он непременно передаст его своим дочерям, потому что каждая дочь наследует только X-хромосому своего отца. Тогда девочки станут носителями, и у каждого из их сыновей будет 50% вероятность развития болезни и так далее.
Хороший способ узнать больше о типе наследования в вашей семье — поговорить с врачом в клиники.
Женщины и МДД
Почему девушки обычно не заболевают МДД? Когда девочка наследует дефектный ген дистрофина от одного из родителей, она обычно также получает здоровый ген дистрофина от своего другого родителя, давая ей достаточно белка, чтобы защитить ее от болезни. Мужчины, которые наследуют мутацию, заболевают, потому что у них нет второго гена дистрофина, который мог бы восполнить неисправный.
В начале эмбрионального развития самки в каждой клетке инактивируется либо Х-хромосома от матери (материнская Х), либо от отца (отцовская Х). Выбор хромосомы для инактивации является случайным. В каждой клетке есть 50-процентная вероятность того, что материнская или отцовская Х-хромосома будет инактивирована, а другая активна.
В большинстве случаев не имеет значения, сколько инактивированных материнских и отцовских Х-хромосом у женщины. Но когда есть мутация в гене Х-хромосомы, например, в гене дистрофина, это имеет большое значение.
Если девушке или женщине приходится полагаться на слишком много Х-хромосом с мутацией гена дистрофина (то есть Х с функциональными генами дистрофина в основном инактивированы), у нее могут развиться симптомы МДД или Беккера.
Обычно, однако, девочки не испытывают полного явления МДД, как мальчики, хотя они все еще имеют симптомы мышечной слабости. У меньшинства женщин с мутацией, называемой проявляющимися носителями, есть некоторые признаки и симптомы мышечной дистрофии Дюшенна.
Для этих женщин дефицит дистрофина может привести к ослаблению мышц спины, ног и рук. У манифестирующих носителей могут быть проблемы с сердцем, которые могут проявляться как одышка или неспособность выполнять умеренные физические упражнения. Проблемы с сердцем, если их не лечить, могут быть довольно серьезными, даже опасными для жизни.
В очень редких случаях у девочки может отсутствовать вторая Х-хромосома полностью, или у ее второй Х может быть серьезное повреждение. В этих случаях она производит мало дистрофина или вообще не вырабатывает его (в зависимости от типа мутации дистрофина), и у нее развивается мышечная дистрофия Дюшенна или мышечная дистрофия Беккера, как у мальчика.
Родственница мальчика с МДД может пройти полный спектр диагностических обследований, чтобы определить её статус носителя. Если выясняется, что она является носителем МДД, регулярные исследования силы и тщательный мониторинг сердечной деятельности могут помочь ей справиться с любыми симптомами, которые могут возникнуть.
Признаки и симптомы мышечной дистрофии Дюшенна
Дети с мышечной дистрофией Дюшенна (МДД) часто поздно начинают ходить.
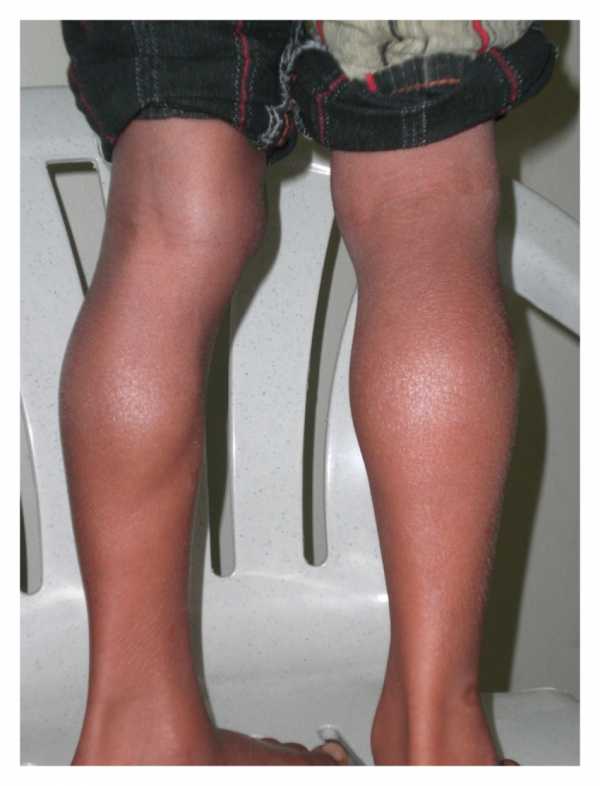
У малышей родители могут заметить увеличение икроножных мышц (см. изображение справа). Это увеличение известно как псевдогипертрофия, или «ложное увеличение» мышц, потому что мышечная ткань является ненормальной и может содержать рубцовую ткань.
Дошкольник с МДД может показаться неуклюжим и часто падать. Родители также могут заметить, что детям трудно подниматься по лестнице, вставать с постели или бегать.
К школьному возрасту детям тяжело ходить и передвигаться, ребенок часто падает. Чтобы сохранить равновесие, они могут выпячивать животы и откидывать плечи. Детям также трудно поднимать руки.
Многие дети с МДД начинают пользоваться инвалидной коляской в возрасте от 7 до 12 лет. Переход на инвалидную коляску обычно происходит постепенно; сначала кресло может потребоваться только для сохранения энергии ребенка при преодолении больших расстояний. (Дети часто вырабатывают новую самостоятельность после полного перехода на инвалидную коляску с электроприводом.)
В подростковом возрасте для действий, связанных с руками, ногами или туловищем, может потребоваться помощь или механическая поддержка.
Боль и чувствительность
Мышечная дистрофия Дюшенна само по себе обычно не болезненна. Некоторые люди иногда сообщают о мышечных судорогах; обычно их можно лечить безрецептурными болеутоляющими средствами.
Поскольку мышечная дистрофия не влияет непосредственно на нервы, осязание и другие чувства остаются нормальными, как и контроль над гладкими или непроизвольными мышцами мочевого пузыря, кишечника и половыми функциями.
Сердце
Недостаток дистрофина может ослабить мышечный слой сердца (миокард), что приводит к состоянию, которое называется кардиомиопатией. Со временем, иногда еще в подростковом возрасте, ущерб, нанесенный МДД сердцу, может стать опасным для жизни. Сердце должно тщательно и постоянно контролироваться, как правило, детским кардиологом.
Дыхательная функция
Начиная примерно с 10-летнего возраста, диафрагма и другие мышцы, управляющие легкими, могут ослабнуть, что сделает легкие менее эффективными при движении воздуха внутрь и наружу. Хотя ребенок может не жаловаться на одышку, к числу проблем и признаков, указывающих на плохое состояние дыхания, относятся головные боли, умственная отсталость, трудности с концентрацией или бессонницей, а также ночные кошмары.
Ослабленные дыхательные мышцы затрудняют кашель, что приводит к повышенному риску серьезной респираторной инфекции. Простая простуда может быстро перерасти в пневмонию. Важно сделать прививку от гриппа, а при возникновении инфекции — получить быстрое лечение.
Неспособностью к обучению
Около треть мальчиков с МДД имеют некоторую степень неспособности к обучению, хотя немногие имеют серьезную умственную отсталость.
Врачи считают, что аномалии дистрофина в мозге могут оказывать незначительное влияние на когнитивные функции и поведение. Проблемы с обучением при МДД возникают в трех основных областях:
- концентрация внимания;
- словесное обучение и память;
- эмоциональное взаимодействие.
Дети с подозрением на нарушение способности к обучению могут быть обследованы психоневрологом-воспитателем или педиатром.
Если диагностирована неспособность к обучению, образовательные и психологические вмешательства могут начаться сразу же. Специалист может назначить упражнения и методы, которые могут помочь улучшить эти области, а специализированные школы могут оказать особую помощь в обучении.
Диагностика
При диагностике любой формы мышечной дистрофии врач обычно начинает с изучения истории болезни пациента и семьи, и проведения физического обследования. История и физическое состояние имеют большое значение для постановки диагноза, даже до того, как будут выполнены какие-либо сложные диагностические обследования.
Анализ уровня КФК
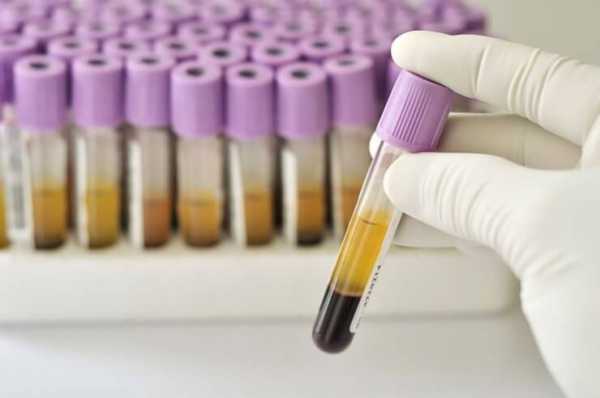
В начале диагностического процесса врачи часто назначают анализ крови, который называется анализом уровня КФК. КФК обозначает креатинфосфокиназу, фермент, который вытекает из поврежденных мышц. Когда в образце крови обнаруживаются повышенные уровни КФК, это обычно означает, что мышцы разрушаются из-за какого-то ненормального процесса, такого как мышечная дистрофия или воспаление.
Очень высокий уровень КФК говорит о том, что сами мышцы (а не нервы, которые их контролируют) являются вероятной причиной слабости, хотя и не говорят точно, каким может быть мышечное расстройство.
Генетическое тестирование
Генетическое тестирование включает анализ ДНК любых клеток (обычно используются клетки крови), чтобы определить, есть ли мутация в гене дистрофина, и если да, то где именно это происходит. Такое тестирование ДНК на мутации дистрофина широко доступно.
Родственники мужчин и мальчиков с МДД могут пройти анализ ДНК, чтобы определить, являются ли они носителями заболевания. Женщины, которые являются носителями МДД, могут передать болезнь своим сыновьям, а статус носителей — своим дочерям. В меньшинстве случаев у девочек и женщин, которые являются носителями МДД, могут у самих проявляться симптомы МДД, такие как мышечная слабость и проблемы с сердцем. Эти симптомы могут не проявляться до зрелого возраста.
Несколько экспериментальных препаратов, которые в настоящее время разрабатываются для лечения мышечной дистрофии Дюшенна, требуют знания точной генетической мутации человека, поэтому генетическое тестирование стало важным не только для диагностики, но и, возможно, для будущего лечения.
Биопсия мышечной ткани
Чтобы получить больше информации, врач может заказать биопсию мышечной ткани, хирургическое взятие небольшого образца мышцы у пациента. Изучив этот образец, врачи могут многое рассказать о том, что на самом деле происходит внутри мышц.
Современные методики могут использовать биопсию, чтобы отличить мышечные дистрофии от воспалительных и других расстройств, а также для различения различных форм мышечной дистрофии. Например, количество функционального белка дистрофина, обнаруженного в образце мышечной биопсии, проливает свет на то, может ли течение болезни быть МДД (без дистрофина) или более умеренная мышечная дистрофия Беккера (с некоторым частично присутствующим дистрофином).
Лечение мышечной дистрофии Дюшенна
Благодаря достижениям во многих областях медицины, таких как кардиология и пульмонология, люди с мышечной дистрофией Дюшенна в 21-м веке живут дольше, чем в предыдущие десятилетия, часто в зрелом возрасте.
Использование доступных процедур может помочь сохранить комфорт и функционирование и продлить жизнь.
Лекарственные препараты

Лекарства, которые уменьшают нагрузку на сердце, иногда назначают и при МДД.
Было обнаружено, что лекарства, принадлежащие к группе, известной как кортикостероиды, эффективны для замедления развития МДД. Кортикостероиды, в частности такие препараты как Преднизон и Дефлазакорт бывают полезны при лечении МДД.
Несколько проведенных исследований этих препаратов при мышечной дистрофии Дюшенна показали значительное увеличение силы, функции мышц по времени (например, время, необходимое для подъема по лестнице) и функции легких.
Постоянное использование кортикостероидов является частью лечения МДД, однако такое лечение может привести к побочным эффектам, а быстрая отмена кортикостероидов может привести к опасным для жизни осложнениям.
Физиотерапия и трудотерапия
Программа физиотерапии является частью лечения МДД. Врач направит вас к физиотерапевту для тщательной оценки и рекомендаций. Основными задачами физиотерапии являются увеличение подвижности суставов и предотвращение контрактур и сколиоза.
В то время как физиотерапия подчеркивает мобильность и, где возможно, укрепление больших групп мышц, трудотерапия фокусируется на конкретных видах деятельности и функциях. Трудотерапия может помочь в выполнении заданий по работе, отдыху или повседневной жизни, таких как переодевание или использование компьютера.
Подтяжки, стоячие рамы и инвалидные коляски
Подтяжки, также называемые ортезами, поддерживают лодыжку и стопу или могут надеваться выше колена.
Ортезы на голеностопный сустав иногда назначают для ночного ношения, чтобы нога не смотрела вниз и не растягивала ахиллово сухожилие во время сна ребенка.
Ношение ортезов в течение нескольких часов каждый день, даже при минимальном весе, способствует лучшему кровообращению, здоровью костей и прямому позвоночнику.
Рано или поздно, при МДД понадобиться инвалидная коляска, как правило, примерно к 12 годам жизни. Если нет травмы, перелома ноги, использование инвалидной коляски обычно происходит постепенно. Многие сначала используют инвалидные коляски при передвижениях на большие расстояния, например в школе или торговом центре.

Хотя ребенок и родители могут бояться инвалидного кресла как символа инвалидности, большинство из них считают, что, когда начинают им пользоваться, дети на самом деле более самостоятельны, энергичны и независимы, чем при попытке ходить.
Есть также и другие средства передвижения и помощи, которые могут облегчить жизнь родителей и опекунов. Одним из самых простых способов помощи является использование специализированной доски для переноса, которая помогает человеку садиться в инвалидную коляску и вставать из нее.
Механические подъемники, кресла для душа и электронные кровати также могут быть полезны.
Упражнения
Упражнения могут помочь нарастить скелетные мышцы, сохранить сердечно-сосудистую систему здоровой и улучшить самочувствие. Но при мышечной дистрофии слишком много упражнений может повредить мышцы. Проконсультируйтесь с врачом о том, сколько упражнений лучше делать. Человек с МДД может осуществлять умеренное количество упражнений, но не должен истощать себя.
Многие эксперты рекомендуют плавание и водные упражнения (водную терапию) как хороший способ поддерживать мышцы в тонусе, насколько это возможно, не вызывая чрезмерного напряжения на них. Вода помогает защитить от определенных видов мышечного напряжения и травм.
Перед выполнением любой программы упражнений обязательно проведите кардиологическое обследование.
Контрактура суставов
Воздействие мышечной дистрофии Дюшенна может быть значительно сведено к минимуму, если держать тело максимально гибким, вертикальным и подвижным. Есть несколько способов сделать это.
По мере разрушения мышц у человека с мышечной дистрофией часто развиваются состояние в суставах, известная как контрактура (ограничение пассивных движений в суставе). Если их не лечить, они станут серьезными, вызывая дискомфорт и ограничивая подвижность и гибкость. Контрактуры могут поражать колени, бедра, ступни, локти, запястья и пальцы.
Однако есть много способов минимизировать и отложить контрактуру. Упражнения с диапазоном движения, выполняемые по регулярному графику, помогают задержать контрактуры, предотвращая преждевременное сокращение сухожилий. Очень важно, чтобы физиотерапевт показал вам, как правильно выполнять упражнения с диапазоном движений.
Крепления (опоры) на нижних конечностях также могут помочь сохранить растянутые и гибкие конечности, задерживая начало контрактуры.
Когда контрактура осложнилась, может быть выполнена операция, чтобы ослабить её. Процедура высвобождения сухожилия, также называемая операцией пяточной кости, часто проводится для лечения голеностопного сустава и других контрактур, когда ребенок все еще ходит. Обычно после этого мальчику нужно будет носить нижние опоры.
Побочные реакции на обезболивающие
Люди с МДД могут иметь неожиданные побочные реакции на определенные виды анестезии. Важно, чтобы при проведении любых операций хирург знала о МДД пациента, чтобы можно было избежать осложнений или быстро их лечить.
Искривление позвоночника
У молодых мужчин с МДД позвоночник может постепенно вытягиваться в изогнутую форму. Позвоночник может изгибаться из стороны в сторону (сколиоз) или вперед в форме «горбуна» (кифоз).
Сколиоз обычно появляется после того, как мальчик начинает пользоваться инвалидной коляской. Искривление, которое иногда наблюдается у тех, кто все еще ходит, называется лордозом.
Тяжелый сколиоз может мешать сидению, сну и даже дыханию, поэтому следует принять меры, чтобы предотвратить его возникновения. Обычно назначаются упражнения.
Вылечить сколиоз и прочие искривления также может операция по выпрямлению позвоночника, при которой в позвоночник вставляется металлические стержни с крючками. Операция для детей с МДД обычно проводится в подростковом возрасте.
как вернуть исчезающие мышцы? — Рамблер/новости
СодержаниеЧто такое мышечная дистрофия Дюшенна?Стандартные методы леченияЛекарства при миодистрофии ДюшеннаРедактирование генома и технология CRISPR/CasДругие исследования: искусственная хромосома, вирусы и мутации
7 сентября — международный день распространения информации о синдроме Дюшенна. Это одна из самых часто встречающихся наследственных генетических патологий — диагностируется она у одного из 3000 новорожденных мальчиков. Пока что способов полного излечения от болезни не существует, но новые разработки дают шанс изменить ситуацию. Какой может быть терапия будущего, расскажет MedAboutMe.
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна — наследственная генетическая патология, одна из тех, которые поражают только мальчиков. Болезнь передается от матерей, но сами женщины от нее не страдают, а являются здоровыми носителями пораженного гена. Ген может передаваться по женской линии многие поколения и никак не проявляться, поэтому рождение ребенка с дистрофией Дюшенна для семьи часто становится неожиданностью.
Патология заключена в гене, кодирующем белок дистрофин — при болезни он вырабатывается в недостаточном количестве или вовсе отсутствует. А поскольку дистрофин является основой мышечных волокон, у детей с дистрофией Дюшенна мышцы постепенно ослабевают, перерождаются и заменяются жировой или соединительной тканью. Это приводит к инвалидизации — болезнь неуклонно прогрессирует, постепенно захватывает все больше мышц.
Даже с существующим сегодня лечением мальчики с миодистрофией Дюшенна живут в среднем 20-25 лет. В некоторых случаях больные доживают до 40, но пока это все же исключение, чем правило.
Первые симптомы болезни возникают еще до 3 лет, в этом возрасте у ребенка проявляется:
быстрая утомляемость; отставание в развитии; трудности с освоением навыка ходьбы, дети часто ходят на пальчиках; симптом Говерса — при попытке встать ребенок активно помогает себе руками, поскольку мышцы ног уже не справляются.
Болезнь прогрессирует, и уже у больных 15-18 лет наблюдаются следующие признаки:
деформация скелета; неспособность передвигаться без инвалидного кресла, вставать, самостоятельно сидеть, иногда даже двигать руками; эндокринные расстройства отмечаются у 30-50% больных; поражение сердечной мышцы, кардиомиопатия; поражение дыхательной мускулатуры; иногда встречаются нарушения психического развития.
На том этапе, когда болезнь захватывает сердце и легкие, помочь пациенту уже практически невозможно.
Стандартные методы лечения
Болезнь была описана еще в XIX веке неврологом Гийомом Дюшенном, в честь которого она и названа. И с того времени врачи ищут эффективные способы терапии. Первым методом, который используется до сих пор, стало применение ортопедических устройств.
Тренировка мышц помогает немного замедлить процесс их разрушения. Для пациентов применяется как активная (пока они сами в состоянии заниматься), так и пассивная тренировка. Врачи отмечают, что регулярные упражнения способны продлить период, когда пациент передвигается без инвалидной коляски.
По статистке, практически половина больных миодистрофией переживают операцию на позвоночнике. При слабых мышцах скелет искривляется, что существенно ухудшает течение болезни. Поэтому на помощь таким больным приходят различные ортопедические приспособления — фиксация костей и суставов позволяет замедлить их деформацию.
Современные разработки в этом направлении терапии давно вышли за пределы простых поддерживающих средств. Так, разработанная нидерландским Университетом Твенте роботизированная рука A-Gear способна заменить человеку атрофированную конечность. Искусственная рука улавливает электрические сигналы от мышц или минимальное мышечное напряжение. Люди с миодистрофией Дюшенна могут управлять механической конечностью, движения при этом получаются очень точными и естественными. «К исследованию мы привлекли много участников, которые уже на протяжении 3-5 лет утратили способность двигать руками, — говорит ученый Джоан Лобо-Прат (Joan Lobo-Prat). — A-Gear помогла им частично вернуть функциональность конечностей».
Лекарства при миодистрофии Дюшенна
Медикаментозное лечение является важной частью общей терапии. Наиболее популярной группой препаратов для пациентов с миодистрофией Дюшенна являются стероиды: преднизолон, дефлазакорт и другие. Начинают их прием еще в возрасте 4-6 лет, когда проявляются признаки болезни, но существенного ухудшения состояния нет. Стероиды помогают отсрочить развитие тяжелых симптомов, но в долгосрочной перспективе они неэффективны. Как только у пациента начинает прогрессировать атрофия, улучшить состояние мышц эти лекарства уже не могут. И все же стероиды остаются важной составляющей лечения дистрофии Дюшенна.
Существуют и другие разработки, направленные на улучшение работы мышц. Так, в 2016 году FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) одобрило лекарство Exondys51 (этиплирсен), которое сможет стать конкурентом стероидам. По данным проведенных исследований, вещество способно укрепить мышечную ткань, ведь оно увеличивает уровень дистрофина в скелетных мышцах. Однако не все врачи разделяют мнение FDA, поскольку утверждение препарата проходило по ускоренной процедуре. По факту сейчас нет однозначных доказательств, что он может отсрочить развитие паралича или ослабить симптомы развитого заболевания. А значит, Exondys51, как и стероиды, проявляет эффективность лишь в коротком периоде болезни. Несмотря на то, что лекарство уже утверждено, FDA продолжает исследования. Если эффективность препарата не будет подтверждена, его производство остановят.
Европейский Комитет по лекарственным препаратам для человека (CHMP) в 2014 году зарегистрировал другой медикамент — Трансларна (аталурен). В отличие от других лекарств, которые только укрепляют мышцы, аталурен способен восстанавливать нарушенный синтез белков. А значит, теоретически устраняет саму причину развития болезни. Поскольку это новая разработка, понять, насколько она эффективна в долгосрочной перспективе, пока трудно. Но промежуточные результаты достаточно обнадеживающие.
Редактирование генома и технология CRISPR/Cas Миодистрофия Дюшенна вызвана патологией в гене, поэтому любое медикаментозное или физиотерапевтическое лечение является всего лишь симптоматическим. Вылечить человека можно только с помощью генной терапии — редактирования кода ДНК и изменения «больного» гена. Несмотря на то, что пока такое лечение не применяется, новые технологии дают шанс на решение проблемы в будущем.
В последнее время особую популярность получила технология CRISPR/Cas9. В ее основе — действие специального фермента Cas9. Он внедряется в клетку и там способен не только распознавать конкретный участок ДНК-цепочки, но и удалять его. Поскольку врачи знают, какой ген виноват в развитии болезни и где конкретно он находится, с помощью технологии они смогут просто вырезать фрагмент ДНК с патологией. А это принесет полное выздоровление.
Однако недавние исследования выявили ряд трудностей с использованием этой системы. После проведенного эксперимента на эмбрионах оказалось, что метод действительно позволил вылечить до 30% клеток. Но при этом такое внедрение породило большое количество других мутаций.
Возможности CRISPR/Cas9 активно изучают многие лаборатории мира. Положительные результаты, в частности, получили ученые из Юго-западного медицинского центра Техасского университета. Здесь CRISPR/Cas9 успешно применили для лечения мышей с миодистрофией Дюшенна. Также была разработана более точная технология, подходящая именно для этой болезни — в ней белок Cas9 был заменен белком Cpf1. «Мы взяли у пациентов с дистрофией Дюшенна клетки и смогли исправить их в лабораторных условиях, — говорит доктор Эрик Олсон. — После применения CRISPR/Cpf1 в клетках восстановилось производство дистрофина».
Другие исследования: искусственная хромосома, вирусы и мутации
Группа японских и британских ученых предложила еще один альтернативный подход для лечения миодистрофии Дюшенна — внедрение искусственных здоровых хромосом (ген с патологией находится в Х-хромосоме 23-й пары). Такая хромосома будет помещена в стволовую клетку, а позже введена в организм и поможет восстановить мышцы. Пока положительные результаты получены в опытах на мышах. Похожие исследования проводились и в России, в Институте стволовых клеток человека.Еще одно возможное направление в лечении болезни Дюшенна появилось благодаря собаке. Более десяти лет назад ученые из Университета Сан-Паулу в Бразилии специально для изучения выводили щенков с мышечной дистрофией Дюшенна. Однако, несмотря на подтвержденный анализами диагноз, у одного из них, пса Ринго, заболевание не развилось, более того, животное прожило 11 лет без каких-либо симптомов. Ученые связывают это с наличием у собаки другой патологии — мутации в гене Jagged1. «Мы точно не знаем, какова должна быть концентрация белка, который кодируется геном Jagged1, в мышцах, чтобы эффективно защищать от развития болезни, — рассказывает генетик Луис Канкел. — Но история Ринго наталкивает на мысль, что мутация Jagged1 компенсирует атрофию, вызванную нехваткой дистрофина».
Одной из самых обнадеживающих перспектив является масштабное исследование, в котором примут участие реальные пациенты. Суть эксперимента — попробовать внедрить в клетку исправленную копию гена дистрофина. И сделать это планируется с помощью вирусов — именно они станут переносчиками нужного фрагмента ДНК. Испытания будут проводиться в США уже в конце 2017-начале 2018 года. В частности, в исследованиях будет участвовать фармацевтическая компания Pfizer. Пока отобрано 12 детей разных возрастных групп, которым в детской больнице Nationwide штата Огайо будут введены вирусные частицы.Миопатия Дюшенна (прогрессирующая мышечная дистрофия): симптомы, лечение, последствия
Миопатия Дюшенна – серьезное заболевание, которое характеризуется первичной дистрофией мышц.
Развивается данная патология с детских лет и может спровоцировать летальный исход до 25 лет, поэтому требует своевременной диагностики и лечения. Самостоятельно побороть заболевание невозможно, единственное, что может сделать пациент, так это придерживаться рекомендаций специалиста и проводить профилактические мероприятия.
Характеристика заболевания
Заболевание носит название своего первооткрывателя – Дюшена, который смог доказать генетическую природу. Миопатия Дюшена не

Вставание лесенкой — характерный признак синдрома Дюшенна
относится к распространенным патологиям, так как встречается один случай на 3,5 миллиона рожденных детей. Диагностируется исключительно у мужской половины населения в возрасте от 1,5 до 3 лет. Заболевание быстро прогрессирует.
При развитии синдрома происходит процесс гипотрофии мышц, который имеет восходящий характер. В первую очередь затрагиваются мышцы таза и проксимальный отдел ног, затем патология начинает распространяться на мышцы спины и плечи, завершающая стадия затрагивает верхние конечности.
Но стоит учитывать и тот факт, что заболевание оказывает негативное влияние и на иные отделы тела:
- деформация позвоночника;
- наблюдается искривление стоп и грудной клетки;
- заболевания сердца.
- у пациента наблюдается дебильность, которая затрагивает третью часть больных.
Что касается прогноза при миодистрофии Дюшенна, то он не очень благоприятный, так как наблюдается интенсивное прогрессирование синдрома.
Ребенок постепенно перестает ходить к десятилетнему возрасту. Летальный исход наступает в результате инфицирования дыхательных путей или остановки сердца.

Тип наследования
Синдром Дюшена относится к заболеваниям, которые передаются по наследству. К носителям патогенного гена относятся женщины.
Наследуется по рецессивному типу, сцепленному с Х-хромосомой. Большинство случаев, при которых была выявлена миопатия, были спровоцированы новыми мутационными генами. Данный вид заболевания относится к быстропрогрессирующему и злокачественному.
Клиника синдрома
Миопатия Дюшенна начинает проявляться у мальчиков в возрасте с 1,5 лет, к первым симптомам относятся:
- нарушения двигательной функции, пациенту сложно стоять, он испытывает неловкость;
- мальчик часто спотыкается, падает во время прогулок, как следствие развивается двигательная пассивность;
- ребенку составляет трудность подняться вверх по лестнице, походка похоже на «утиную»;
- ребенку тяжело подниматься с кровати или со стула;
- наблюдается гипертрофия икроножных мышц, данный признак относится к наиболее яркому, который сразу свидетельствует о прогрессировании заболевания;
- заболевания сердца;
- нарушения в биоптатах мышц скелета;
- контрактура крупных суставов, деформация стопы наблюдается у пациента в результате интенсивного развития патологии;
- в возрасте десяти лет ребенок перестает передвигаться самостоятельно и становиться прикованным к инвалидному креслу;
- в возрасте 14 лет ребенок становиться полным инвалидом.
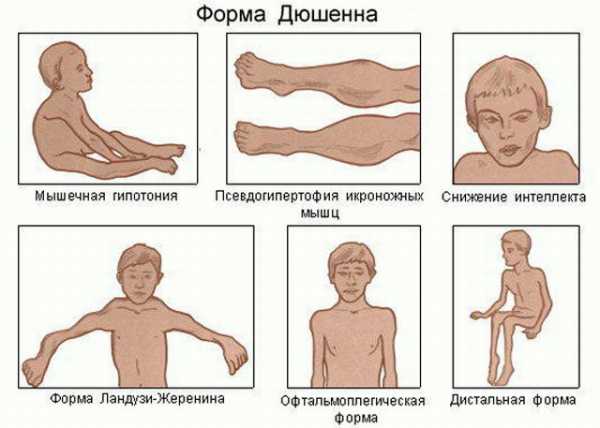
Симптомы миодистрофии Дюшена
Если были выявлена выше представленная симптоматика стоит немедленно пройти обследование и лечение. В данной ситуации не стоит тратить ни минуты, так как от этого зависит жизнь пациента.
Постановка диагноза
В первую очередь пациента осматривают, чтобы выявить внешние проявления болезни Дюшенна. Затем ребенка направляют на дополнительное обследование, которое заключается в следующем:
- электрокардиограмма помогает выявить поражение миокарды латеральное и заднее-нижней стенок левого желудочка;
- определение уровня дистрофина в ткани мышц;
- биохимическое исследование крови, помогает выявить активность фермента креатинфорсфокиназы;
- в обязательном порядке проводиться генетическая диагностика;
- проводиться электронейромиография, помогает выявить некроз мышечной ткани;
- биопсия мышечной ткани, считается главным методом диагностирования миопатии.
На основании полученных результатов ставиться окончательный диагноз, и назначается терапия.
Лечение: цели, методы, трудности
Лечение миопатии Дюшенна невозможно, можно только продлить время жизни и поддержать состояние пациента на стабильном уровне.
Различные методы современной медицины помогают облегчить состояние, замедлить прогрессирование болезни. Ниже будут представлено лечение миодистрофии Дюшена для разных возрастов, но бывают такие случаи, когда требуется применять сразу несколько способов терапии:
- Пациент в возрасте до 5 лет. В данной ситуации не требуется радикальное лечение. Рекомендуется осведомить родителей о заболевании, рассказать о проблемах и последствиях, рассказать, какие нагрузки допустимы для ребенка, и провести в обязательном порядке генетическую консультацию.
- Пациент в возрасте до 8 лет. Большинству детей в таком возрасте уже требуется поддержка мускулатуры нижних конечностей. Кортикостероиды помогают замедлить ход развития заболевания на некоторое время. Рекомендуется принимать Преднизолон или Дефлазакорт.
- Пациент в возрасте от 8 лет до 20 лет. Наблюдается постепенное ослабление мышц, которое в результате заставляет ребенка прибегнуть к инвалидному креслу.
- Пациент в возрасте от 20 лет. Препараты в данной ситуации помогут только облегчить состояние, начинает прогрессировать инфекция дыхательных путей.
В течение всей жизни пациент должен принимать препараты, которые обеспечивают поддержку организма:
- витамины В и Е, аминокислоты, кальций, анаболические гормоны, калия оротат;
- Прозерином, Галантаминон, Оксазилон.
Также курсами проводят инъекции Ретаболила, АТФ, Цереброзилил, Анарилин. Рекомендуется принимать глютаминовую кислоту, Ексазил.
 Параллельно проводиться лечение ЛФК, которое осуществляется непродолжительными курсами с перерывами. Также рекомендуется делать массаж, электрофорез Прозерина, Липазы, кальция хлорида, применяют лечебные ванны и индуктотермию. В особо тяжелых случаях лечение проводиться в домашних условиях. Чтобы продлить жизнь на несколько лет можно попробовать глюкокортикоидное лечение.
Параллельно проводиться лечение ЛФК, которое осуществляется непродолжительными курсами с перерывами. Также рекомендуется делать массаж, электрофорез Прозерина, Липазы, кальция хлорида, применяют лечебные ванны и индуктотермию. В особо тяжелых случаях лечение проводиться в домашних условиях. Чтобы продлить жизнь на несколько лет можно попробовать глюкокортикоидное лечение.
Параллельно пациент должен постоянно находиться под наблюдением кардиолога. Также обязательно ребенок должен полноценно питаться. В рацион включены: растительные жиры и белковая пища, свежие или приготовленные на пару овощи, фрукты, молочные продукты, овсянка, яйца, мед, орехи и морковь. Не стоит потреблять крепкий чай или кофе, спиртные напитки, приправы, сахар, капусту и картофель.
Осложнения и последствия
Как уже говорилось ранее, данное заболевание сильно укорачивает жизнь человека и это можно считать самым серьезным последствием. Так как патология быстро прогрессирует, то мышцы становятся слабыми, возникают необратимые последствия в работе сердца, наблюдается сбой дыхательной системы.
Если миопатия Дюшена будет своевременно выявлена и лечение будет подобрано правильно, то пациент сможет дожить до 30 лет.
К осложнениям можно отнести такие заболевания и патологии:
- Остеопороз. Чтобы постараться отсрочить данную патологии рекомендуется принимать витамин Д и кальций, поэтому стоит скорректировать питание. При остеопорозе принимают биосфосфонаты.
- Заболевания суставов и позвоночника. Суставы становятся практически полностью подвижными, поэтому рекомендуется носить шины. В некоторых случаях проводят операции.
- Заболевания пищеварительной системы и проблемы с весом. С возрастом человек начинает терять в весе из-за некроза
 мышц, поэтому требуется постоянная консультация диетолога. Также пациента может потревожить и запор, который также является следствием малоподвижного образа жизни. В данной ситуации рекомендуется принимать слабительные препараты, больше есть продуктов, которые богаты на клетчатку. В возрасте от 17 лет у больного могут возникнуть проблемы с жевательной и глотательной функцией. В редких случаях проводиться гатростомия.
мышц, поэтому требуется постоянная консультация диетолога. Также пациента может потревожить и запор, который также является следствием малоподвижного образа жизни. В данной ситуации рекомендуется принимать слабительные препараты, больше есть продуктов, которые богаты на клетчатку. В возрасте от 17 лет у больного могут возникнуть проблемы с жевательной и глотательной функцией. В редких случаях проводиться гатростомия. - Нарушения дыхательной функции. Из-за слабого кашлевого механизма могут прогрессировать респираторные инфекции, так как бактерии и слизь не выводятся. В качестве профилактики рекомендуется делать прививки. Также по мере прогрессирования патологии у пациента наблюдается снижение уровня кислорода в организме, как следствие возникают утренние головные боли, бессонница, слабость, нервная возбудимость, особенно во сне.
- Заболевания сердца. В большинстве случаев развивается кардиомиопатия.
Профилактика болезни затрудненная, так как данная патология является генетической, поэтому нужно уделить внимание консультации у генетиков тем семьям, у которых наблюдается отягощенная наследственность.
фото, причины, симптомы, диагностика, лечение и профилактика
Одной из самых грозных первичных мышечных дистрофий, которая начинается в раннем детстве и приводит к летальному исходу до достижения больным 25-летнего возраста, является миопатия Дюшена (полное название – прогрессирующая мышечная дистрофия Дюшена).
Краткая характеристика миопатии Дюшена
Заболевание было впервые описано в 1868 году Дюшеном (Duchenne) и является генетическим. Причем миопатия Дюшена имеет общую, генетически единую форму с миопатией Беккера, однако отличается целым рядом клинических признаков.
Встречается болезнь у одного из 3-3,5 тысяч новорожденных мальчиков и обнаруживается в возрасте 1,5-3 лет и быстро прогрессирует.
Обычно больные не доживают даже до 30-летнего возраста (по некоторым данным, и вовсе многие умирают в возрасте 20-22 лет).
Распространению процесса гипотрофии мышц свойственен восходящий характер:
- Сначала в процесс вовлекаются мышцы тазового пояса, а также мышцы проксимальных отделов ног (нижних конечностей),
- Затем страдают мышцы спины и плечевого пояса,
- После этого очередь доходит до проксимальных отделов рук (верхних конечностей).
Уже в самом начале заболевания угасают или значительно снижаются коленные рефлексы, при этом сухожильные рефлексы рук и Ахиллов рефлекс сохраняются ещё очень длительное время.
Другие признаки этого заболевания:
- Прогрессирование кифоза, лордоза или других вторичных деформаций позвоночника,
- Искривление грудной клетки (она становится килевидной или седловидной), стоп,
- Развиваются ретракции сухожилий на фоне контрактур в суставах.
- Очень часто при миопатии Дюшена наблюдаются проблемы с сердцем, а именно кардиомиопатия, симптомами которой становятся аритмия и гипертрофия левого желудочка.
- Хотя обычно при миопатиях интеллект не страдает, в этом случае у 25-30% пациентов наблюдается олигофрения (а именно, в степени дебильности). У остальных больных интеллект сохранен.
Прогноз заболевания неблагоприятный – миодистрофия Дюшена быстро прогрессирует, больные теряют способность к самостоятельной ходьбе после 10-12 лет, а умирают в молодом возрасте по причине интеркуррентных инфекций (дыхательной недостаточности) или от сердечной недостаточности.
Фото миопатии Дюшена: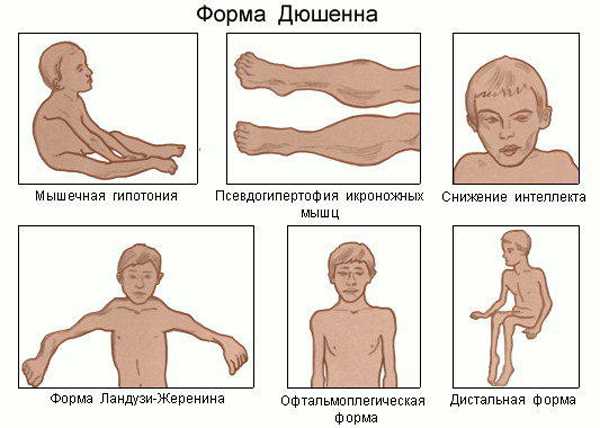
Причины
Миопатия Дюшена является наследственным заболеванием, причем носителями его гена являются представительницы женского пола. Наследуется эта миопатия по рецессивному типу, сцепленному с Х-хромосомой. Более того, около трети всех случаев выявления миопатии Дюшена вызваны новыми мутациями генов. Болеют исключительно мальчики.
Из всего многообразия миопия эта является наиболее злокачественной и быстро прогрессирующей.
Хотя миопатия Дюшена является наследственным заболеванием, в 30% случаев причиной является генная мутация.
Симптомы заболевания
Заболевание начинает проявляться в возрасте 1,5-5 лет, его первыми признаками являются:
- Неустойчивость, двигательная неловкость.
- Постоянные спотыкания и падения во время ходьбы, что развивает у ребенка страх перед ходьбой, чем обусловлена двигательная пассивность.
- Ребенку сложно подниматься по лестнице, а походка становится вразвалочку, «утиной».
- Также затруднительным становится подъем из положения лежа или сидя – ребенок прибегает к так называемым «приемам Гроверса» — это «взбирание по самому себе» и «взбирание лесенкой».
- Яркий признак миопатии Дюшена – мнимая гипертрофия мышц, особенно икроножных: на самом деле происходит не развитие мышц, а их перерождение в жировую и соединительную ткани.
- Как уже упоминалось, одним из симптомов миопатии Дюшена является поражение сердца, причиной которого, по мнению зарубежных исследователей, становится недостаток в кардиомиоцитах дистрофина.
- Наблюдаются признаки миопатии в биоптатах мышц скелета.
- Со временем, по мере прогрессирования мышечной дистрофии развиваются контрактуры крупных суставов, наблюдается эквиноварусная деформация стопы.
- Ближе к 10-12 годам ребенок уже не может передвигаться самостоятельно и вынужден пользоваться инвалидным креслом.
- К 15 годам развивается глубокая инвалидизация больного.
Знаете ли вы, какие бывают симптомы серотонинового синдрома? Причины и последствия.
Всё о принципах лечения миофасциального болевого синдрома можно найти здесь.
Диагностика
 Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
- На ЭКГ выявляется поражение миокарда латеральной и задне-нижней стенок левого желудочка, что определяется по следующим показателям: высокий зубец наблюдается в отведении V6; глубокий зубец Q наблюдается в отведениях V6, aVF, 2 и 3.
- Также исследуется содержание дистрофина в мышечной ткани (при этом заболевании дистрофия не выявляется).
- В ходе биохимических исследований в плазме крови определяется активность КФК (фермента креатинфорсфокиназы), которая обычно существенно повышена (в том числе у носительниц гена). Иногда для уточнения источника исследуются изоферменты КФК.
- Проводится также генодиагностика.
- Фибрилляции на ЭМГ сообщают о некрозе мышечных волокон.
- Биопсия мышц является одним из основных методов диагностики миопатии Дюшена, причем выбирается умеренно пораженная мышца, поскольку очень ослабленная и существенно пострадавшая мышца окажется неинформативной.
Наиболее достоверными являются анализы на активность в сыворотке мышечных ферментов, биопсия мышц и ЭМГ (электромиография).
Лечение миопатии Дюшена
При таком тяжелом и быстро прогрессирующем заболевании лечение малоэффективно, обычно в комплексной поддерживающей терапии применяются следующие препараты:
- Группа препаратов, улучшающих обмен веществ в организме: витамины группы В, Е, аминокислоты, препараты кальция, анаболические гормоны, калия оротат.
- Применяется лечение прозерином, галантамином, оксазилом.
- Лечение проводится курсами и, предпочтительнее, в стационаре: ЛФК (особенно замедляющая образование контрактур), так же, как и пассивное растяжение больных мышц, массаж, электрофорез прозерина, лидазы, кальция хлорида, ванны, индуктотермия. Курс лечения повторяется каждые 6-8 недель, а неподвижных детей предпочтительнее лечить на дому.
- Последние годы популярным стало лечение глюкокортикоидами (по схеме через день), которое продлевает жизнь ещё на несколько лет.
Больному миопатией Дюшена нужно обязательно наблюдаться у врача-кардиолога.
Отдельно стоит отметить важность полноценного питания, в котором обязательно должны присутствовать растительные жиры и белки животного происхождения. Стоит избегать крепкого чая, кофе, алкоголя, пряностей, сахара, капусты и картофеля. В рацион должны входить свежие или приготовленные щадящим способом овощи, фрукты, кисло-молочные продукты, овсяная крупа, яйца, мед, морковь, орехи.
Не секрет, что нервными тиками страдают как взрослые люди, так и дети. Причины нервного тика глаза, диагностика и лечение.
Симптомы алкогольной энцефалопатии хорошо освещены в этой статье.
Лечение нейропатической боли у взрослых зачастую представляет собой длительный процесс и требует комплексного подхода. Подробную информацию об этом заболевании можно получить, перейдя по этой ссылке: http://gidmed.com/bolezni-nevrologii/nevralgija/nejropaticheskaya-bol.html.
Профилактика
Профилактика заболевания затруднена ввиду генетических причин его возникновения, вот почему особо важную роль играет генетическое консультирование тех семей, в которых выявлена отягощенная наследственность.
Новейшие методы и разработки молекулярной генетики помогают достоверно определить природу мутации гена, «вычислить» прогноз его заболевания, но самое главное – такие методы позволяют провести перинатальное обследование в случае повторной беременности.
Миопатия — Википедия
Материал из Википедии — свободной энциклопедии
Миопа́тия (от др.-греч. μῦς — мышца и πάθος — болезнь, страдание) — хронические прогрессирующие нервно-мышечные заболевания, характеризующиеся первичным поражением мышц.
В настоящее время существуют разные классификации. Более ранней была клиническая классификация нервно-мышечных заболеваний, которые ранее все считались болезнью мышц. В рамках этой классификации выделяли конечностно-поясную, лице-плече-лопаточную, гумеро-тибиальную, и другие виды. Чёткой клинической классификации нервно-мышечным заболеваниям и миопатиям в частности сейчас нет, но до сих пор широко используются исторически сложившиеся и закреплённые за некоторыми заболеваниями.
Возникновение патогенетической классификации связано с появлением новых знаний о том, что миодистрофии могут возникать вследствие множественного поражения нервов, вследствие нарушения обменных процессов, токсических воздействий, воспалительных процессов, породило деление на первично-мышечные заболевания, невральные амиотрофии и др. Патогенетическая классификация миопатии с развитием науки становится все детальней, и сейчас учёные стремятся указывать в диагнозе поражённый белок (например, кальпаинопатия, титинопатия и др.). Уточнение дефектного белка позволяет также предположить и установить мутацию.
Необходимо разделять наследственные и приобретённые миопатии. Анамнез наследственных миопатий содержит более или менее чёткие указания на наличие заболевания у родственников, хотя это не обязательно. Примеры наследственных миопатий: дистрофические миопатии (миопатия Дюшенна и др.), митохондриальные миопатии, болезни накопления (болезнь Помпе и др.).
Патогенез различных видов миопатий различен, в зависимости от поражённого гена и даже локуса. Нарушение синтеза структурных белков миофибрилл приводит к появлению дистрофических миопатий (или дистрофические миодистрофии — ДМД. Например, ДМД Дюшенна, конечностно-поясные миодистрофии и др.). Нарушение синтеза или снижение активности ферментов приводят к появлению болезней накопления. В любом случае рано или поздно появятся основные проявления миопатии (как и других нервно-мышечных заболеваний): мышечная слабость и атрофия мышц.
Патогенез различных видов миопатий различен, в зависимости от поражённого гена и даже локуса.
Существует множество форм миопатий, отличающихся возрастом дебюта заболевания, тяжестью течения и преимущественным поражением тех или иных групп мышц. Сложность диагностики некоторых заболеваний заключается в том, что эти заболевания редки, а также в том, что даже в пределах одной нозологии имеется широкая вариабельность симптомов.
Диагноз ставится на основании
- клинических проявлений (слабость мышц, их похудение)
- Данных ЭМГ (миопатический паттерн на игольчатой ЭМГ)
- КФК плазмы крови (повышение до десятков и сотен верхней границ нормы)
- МРТ мышц (картина жирового или соединительно-тканного перерождения мышц, характерное для какого-либо заболевания распределение поражения мышц)
- Биопсия нервно-мышечного лоскута. Более информативно использование криосрезов биоптатов, но и за рубежом чаще, а в России почти всегда используется обычный способ приготовления препаратов. Можно выделить:
- Простая микроскопия с окраской.
- иммуногистохимическое исследование.
- в некоторых случаях необходимо применять специальные анализы крови на наличие определённых ферментов (болезнь Помпе)
В дифференциальный диагноз входят нервно-мышечные заболевания другого генеза (невральные, спинальные мышечные атрофии), травматические поражения мышц и нервов, воспалительные мышечные заболевания, а также широкий дифференциальный диагноз миопатий друг c другом.
В настоящее время радикального лечения таких заболеваний не существует, однако разрабатываются методы генной и клеточной терапии, а для миопатии в рамках болезни накопления — заместительная терапия. В 2014 году Европейский Комитет по лекарственным препаратам для человека (CHMP) разрешил регистрацию препарата Трансларна (Translarna), или аталурен (ataluren), для лечения мышечной дистрофии Дюшена.
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ 1 2 3 Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Беленикин М.С., Жилина С.С., Баринов А.А., Шарина М.Ю., Брюханова Н.О., Магомедова Р.М., Мещерякова Т.И., Петрин А.Н., Демидова И.А., Прокопьев Г.Г., Мутовин Г.Р., Притыко А.Г. Аллельный вариант врожденной миопатии Салиха // Российский вестник перинатологии и педиатрии. — 2015. — № 3. — С. 89-93. — ISSN 1027-4065.