Миотонический синдром у взрослых что это такое
виды, причины, признаки, симптомы и лечение
Миотония – генетически обусловленная патология, относящаяся к категории нервно-мышечных недугов. Примечательно то, что в зависимости от своей разновидности может манифестировать в разном возрасте, а не с момента появления малыша на свет. Основная причина развития болезни состоит в передаче ребёнку мутирующего гена от одного или обоих родителей, при этом они могут быть совершенно здоровыми. Иные предпосылки, влияющие на развитие такого редкого заболевания, остаются неизвестными.
Онлайн консультация по заболеванию «Миотония».
Задайте бесплатно вопрос специалистам: Невролог.Главным клиническим проявлением выступает резкое затруднение расслабления мышц после их длительного и сильного сокращения. Остальная симптоматика обуславливается разновидностью патологии.
Поставить правильный диагноз может врач-невролог на основании не только инструментальных обследований, а также по причине присутствия симптома «кулака», который является главным диагностическим тестом.
Специально предназначенных методов терапии миотонии не разработано. Для снижения выраженности симптоматики обращаются к консервативным методикам. Несмотря на это, прогноз для жизни и трудоспособности – благоприятный.
Этиология
Во всех ситуациях развитие миотонии обуславливается генетическими или хромосомными нарушениями.
Самой распространённой формой считается миотония Томпсона, которая наследуется по аутосомно-доминантному типу. Это означает, что для развития недуга достаточно получения всего лишь одного дефектного гена от кого-либо из родителей.
В редких случаях наблюдается наследование по аутосомно-рецессивному типу. Отличительной чертой является то, что для проявления заболевания нужно по одному мутировавшему гену от каждого родителя. В таких ситуациях болезнь протекает тяжелее и проявляется в раннем возрасте.
Миотония Россолимо-Баттена-Штейнерта-Куршмана – наследственная патология с медленным прогрессированием. Её основа заключается присутствии дефекта в гене девятнадцатой хромосомы. Именно такой ген отвечает за синтезирование миотинин-протеикиназ. Тяжесть протекания такой разновидности поражения мышц диктуется количеством повторов.
Врождённая миотония Беккера может быть унаследована по аутосомно-рецессивному типу и развивается на фоне таких нарушений:
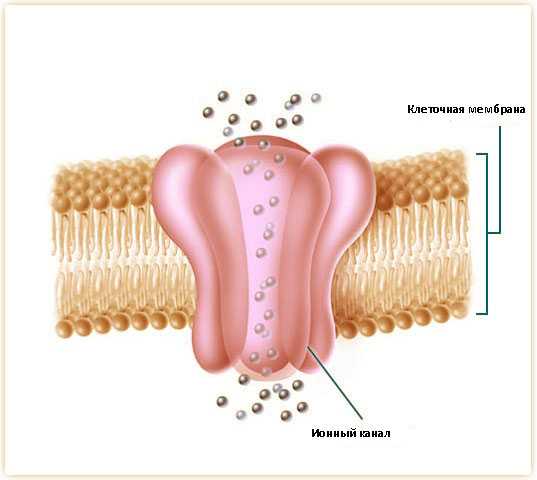
- функциональная неполноценность белка, находящегося в хлорных каналах мышечных волокон мышц скелета;
- нестабильность ионного равновесия;
- дисфункция мембраны волокон, что ведёт к гипервозбудимости.
Врождённая парамиотония Эйленбурга может наследоваться по аутосомно-доминантному характеру и заключается в патологиях натриевых каналов. Симптоматика развивается на фоне:
- повышенной возбудимости мембран мышечных волокон;
- неправильной работы сократительных элементов мышц.
Классификация
Специалистами из области неврологии принято выделять несколько видов подобного заболевания, а именно:
В зависимости от количества тринуклеотидных повторов, различают такие степени тяжести протекания болезни:
- мягкую – частота повторов увеличивается до восьмидесяти;
- среднетяжелую – количество повторений варьируется от ста до 500;
- тяжёлую – частота повторов достигает 2000. Такое проявление наиболее характерно для врождённой формы.
Стоит отметить, что в норме число тринуклеотидных повторов составляет от пяти до тридцати семи.
Симптоматика
Для всех разновидностей нарушения мышечной активности характерно присутствие признака «кулака», который является не только основным клиническим тестом, проводимым во время диагностики, но также выступает в качестве наиболее специфического симптома.
Суть его заключается в том, что человек не способен быстро разжать кулак – чтобы это сделать пациенту с подобным заболеванием необходимы некоторое время и приложение усилий. Примечательно то, что при повторных попытках выполнить подобное движение, миотонические проявления угасают. Единственное исключение составляет миотония Эйленбурга, при которой скованность ещё больше усиливается во время повторных попыток совершить такое движение.
Помимо этого, скованные движения также наблюдаются при:
- открытии рта;
- быстром открытии крепко зажмуренных глаз;
- вставании со стула.
В остальном же каждая разновидность заболевания имеет собственные признаки.
Например, миотония Россолимо-Баттена-Штейнерта-Куршмана характеризуется:
- возникновением первых признаков в возрасте старше пяти лет;
- продолжительность манифестации вплоть до тридцатипятилетнего возраста;
- пиком выраженности симптоматики в возрастной категории от десяти до двадцати лет;
- нарушением функционирования ЦНС и органов, составляющих сердечно-сосудистую систему;
- развитием эндокринных патологий;
- присутствием катаракты;
- выраженностью миотонических спазмов в жевательных мускулах и мышцах кисти, реже в мускулах нижних конечностей и височных мышцах;
- атрофией различных мышц;
- миопатическим парезом гортани;
- расстройством процесса глотания и воспроизведения голоса;
- приступами апноэ во сне;
- понижением интеллекта, вплоть до дебильности;
- нарушением сна.
Проявления миотонии Беккера представлены:
- возникновением симптоматики с самого рождения ребёнка;
- редкой манифестацией у детей старшего возраста – у девочек до двенадцати лет, у мальчиков – до восемнадцати;
- медленным расслаблением мышц после активного движения;
- снижение спастичности при повторных движениях;
- болевыми ощущениями в мышцах;
- поражение проксимальных и дистальных отделов рук и ног.
Миотония Томпсена выражается в:
- миотоническом феномене, характеризующемся длительной мышечной релаксацией после совершения быстрых движений;
- тоническом спазмировании мышц, что делает невозможным быстро осуществить следующее движение;
- поражении мышц кистей и ног, мимической и жевательной мускулатуры;
- утрате равновесия при быстрой ходьбе;
- атлетическом телосложении, к чему приводит диффузная мышечная гипертрофия.
Симптомы миотонии хондродистрофической формы:
- низкорослость;
- дисплазия тазовых костей;
- ригидность суставов;
- скованностью мимики.
Врождённая дистрофическая миотония может выражаться в:
- нарушении ЧСС;
- гиперсомнии;
- нарушении менструального цикла – у слабой половины человечества;
- гипогонадизме и импотенции – у представителей мужского пола;
- обострении миотонического феномена на холоде.
Диагностика
Установление причин возникновения подобной патологии и определение её разновидности требует выполнения целого комплекса диагностических мероприятий, среди которых:
- изучение истории болезни пациента и его ближайших родственников;
- тщательный физикальный осмотр;
- консультирование специалиста по генетике;
- общеклинический анализ и биохимия крови – покажет наличие антител к потенциал-зависимым калиевым каналам и незначительное возрастание активизации КФК, что характерно для дистрофии;
- анализ крови на гормоны;
- ЭМГ – это основная инструментальная методика диагностирования болезни. Во время приступа миотонического феномена разряды будут сопровождаться звуком «пикирующего бомбардировщика», который возникает на фоне введения и перемещения игольчатого электрода;
- ЭКГ и электронейрография;
- ДНК-диагностика.
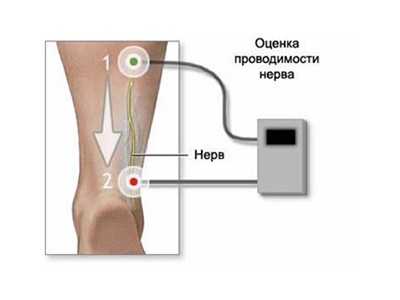
Электронейрография
Дифференцировать миотонию у детей и взрослых необходимо с синдромом «ригидного человека», имеющим схожие признаки и симптоматику.
Лечение
Поскольку врождённая миотония обуславливается генетическими и хромосомными нарушениями, то вылечить её полностью невозможно. Для устранения мышечных дефектов прибегают к таким консервативным методикам:
- соблюдение щадящего рациона питания, направленного на ограничение потребления солей калия;
- прохождение курса лечебного массажа;
- выполнение ЛФК упражнений;
- физиотерапевтические процедуры, среди которых электромиостимуляция;
- избегание переохлаждения, поскольку при некоторых формах протекания на холоде может обостряться симптоматика;
- приём медикаментов, назначаемых лечащим врачом в индивидуальном порядке для каждого пациента.
В редких ситуациях можно достичь ремиссии патологии путём проведения иммуносупрессивной терапии, которая предполагает введение:
- иммуноглобулинов человека;
- преднизолона;
- циклофосфамида.
Возможные осложнения
Отсутствие лечения миотонии может привести к развитию:
Профилактика и прогноз
На фоне того, что заболевание имеет столь специфические этиологические факторы, предупредить его развитие не представляется возможным. Единственной мерой профилактики можно считать прохождение генетического обследования двух родителей перед тем, как заводить ребёнка. Это поможет рассчитать вероятность развития миотонии у малыша.
Что касается прогноза, то в подавляющем большинстве случаев он благоприятный. Крайне редко наступает летальный исход из-за развития внезапной сердечной смерти. Прогноз трудоспособности также положительный – при условии рационального трудоустройства.
причины, симптомы, диагностика и лечение
Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании. Врожденная миотония сопровождается мышечной гипертрофией, дистрофическая миотония, напротив, сопровождается мышечными атрофиями. Диагностика миотонии осуществляется при помощи ЭМГ, ЭНГ и исследования вызванных потенциалов. До настоящего времени радикальная медикаментозная терапия миотонии не разработана. Пациентам проводится симптоматическое и метаболическое лечение, массаж, ЛФК, электростимуляция.
Общие сведения
Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании.
Этиология и патогенез миотонии
Среди двух типов дистрофической миотонии тип 1, ген которого картирован в локусе 19q13, встречается наиболее часто (98%). Как и все типы дистрофической миотонии он передается по наследству по аутосомно-доминантному типу. Основной этиологический фактор дистрофической миотонии 1 типа — увеличением количества тринуклеотидных повторов CTG (до нескольких тысяч). Миотонин-протеинкиназа, кодируемая геном DMPK, присутствует не только в скелетных, но и миокарде, а также ЦНС. Этим и объясняются основные клинические проявления дистрофической миотонии.
Врожденная парамиотония Эйленбурга, связанная с патологией натриевых каналов, передается по аутосомно-доминантному типу. Ген SCN4A картирован в локусе 17q23.1-q25.3. Парамиотонические проявления развиваются в связи с повышенной возбудимостью мембраны мышечных волокон и нарушением функционирования сократительных элементов мышцы.
В отличие от большинства нейромиотоний, являющихся приобретенными заболеваниями, идиопатическая нейромиотония (синдром Исаакса) передается по наследству и встречается наиболее часто. Эффективность введения кураре для купирования мышечных спазмов говорит о неврогенной природе нейромиотонии, однако окончательно патогенез заболевания неясен и сегодня. С выявлением повышенного титра антител к потенциал-зависимым калиевым каналам нейромиотонию стали считать аутоиммунным заболеванием. Об аутоиммунном характере заболевания говорит также наблюдаемая в ряде случаев эффективность плазмафереза.
Классификация миотонии
Наиболее распространенные формы миотонии:
- дистрофическая (двух типов)
- хондродистрофическая
- конгенитальная аутосомно-доминантная
- конгенитальная аутосомно-рецессивная
- парамиотония Эйленбурга
- нейромиотония
Клиническая картина миотонии
Симптом «кулака» — основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов — миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика», возникающие при введении и перемещении игольчатого электрода.
Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. Дистрофическая миотония — мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). Для парамиотонии типична т. н. «холодовая миотония» — возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Диагностика миотонии
Дебют дистрофической миотонии приходится, как правило, на юношеский либо взрослый возраст, степень прогрессирования заболевания зависит от генетического дефекта, поэтому тщательный сбор семейного анамнеза имеет большое значение в диагностировании дистрофической миотонии. Миотония Беккера дебютирует в 5-12 лет и характеризуется медленным течением, а миотония Томсена может дебютировать как в детском, так и в зрелом возрасте и протекает, как правило, тяжело и с осложнениями. Физикальное обследование при дистрофической миотонии выявляет атрофию мышц и снижение их силы. Для дистрофической миотонии типа 1 характерна мышечная слабость в дистальных отделах конечностей, для дистрофической миотонии типа 2 — в проксимальных.
С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови.
Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды — патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Для врожденной миотонии типично сохранение параметров ДЕ в пределах нормы, для дистрофической миотонии — сочетание невропатических и миопатических черт. Для диагностики парамиотонии проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества CTG-повторов в гене DMPK.
Дифференциальный диагноз
Как правило, дифференцировать врожденную миотонию от дистрофической миотонии неврологу удается по клиническим признакам. Однако в ряде случаев врожденной миотонии определяют легкую слабость дистальных мышц рук и слабую активность при ЭМГ — признаки, типичные для дистрофической миотонии. Постоянная мышечная активность — клинический признак нейромиотонии — входит в состав синдрома "ригидного человека" (stiff-man syndrome), однако в отличие от нейромиотонии мышечная активность при синдроме "ригидного человека" снижается после введения диазепама, а также во время сна.
Лечение миотонии
Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии — снижение выраженности миотонических проявлений. Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции. Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин, а для снижения уровня калия — диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека, преднизолон, циклофосфамид.
Прогноз при миотонии
Прогноз для жизни при миотонии в целом благоприятный за исключением редких случаев дистрофической миотонии типа 1, когда возможно наступление внезапной сердечной смерти по причине кардиальной патологии. Прогноз для трудоспособности пациентов с врожденными миопатиями также благоприятен (при рациональном трудоустройстве).
Миотонический синдром
Миотонический синдром – это нервно-мышечное нарушение, проявляющееся чаще у детей, нежели у взрослых, и выражающее себя в существенно затрудненном расслаблении мышц после их сокращения. Синдром нередко носит генетический характер, являясь геномной патологией аутосомно-доминантного или аутосомно-рецессивного типа наследования.
Проявления:
- Мышечная слабость, которая выражается в неспособности ребенка держать спинку, прямо сидеть или стоять;
- Чрезвычайно быстрая утомляемость: малыш способен устать при малейшем усилии;
- Апатия, нежелание играть, особенно в активные подвижные игры, безразличие к хобби, характерным для детского возраста;
- Нарушения работы пищеварительного тракта, постоянные запоры и колики;
- Недержание мочи при смехе, чихании, кашле и натуживании;
- Хронические головные боли неясного генеза;
- Визуальные нарушения осанки;
- Стремительное развитие близорукости;
- Речевые расстройства в совокупности;
- Беспричинные и частые потери равновесия;
- Падения при быстрой ходьбе, беге, подъеме по крутым склонам и лестницам;
- Снижение интеллектуальных способностей.
Вы можете самостоятельно идентифицировать синдром следующим образом: легонько постукивайте по икроножной мышце ребенка. В этот момент должен произойти достаточно болезненный спазм, при котором вы явно увидите проявленные мышечные валики.
Их контрастность может сохраняться еще несколько минут после расслабления. Если вы увидели некоторые перечисленные явления в своем ребенке, немедленно покажите его врачу!
Ложное убеждение «Само пройдёт»
Лечение обнаруженного миотонического синдрома следует начать как можно раньше, ведь если игнорировать тревожные признаки, можно нанести вред дальнейшему развитию малыша. Во-первых, он станет позже держать головку и спинку. Во-вторых, значительно нарушится его осанка. И главное, ребенок начнет позже говорить, ведь тоническое напряжение касается и лицевых мышц! Речь больных детей обычно невнятна и медленна, их монолог трудно понять, и еще труднее обучить малыша буквам и словам. Если относительно своего ребенка вы услышали такой диагноз, как миотонический синдром, в срочном порядке запишитесь на консультацию, для адекватной терапии расстройства!Ведь осложнения могут быть действительно печальными: хронический энурез, близорукость, перманентные головные боли, остеохондроз и сколиоз – далеко не полный перечень распространенных «побочных» эффектов нелеченного недуга.
Лечение миотонического синдрома у маленьких детей часто включает в себя массаж и физиотерапевтические процедуры. Комплексные лечебные меры направлены на нормализацию тонуса мышц и нейтрализацию их тонического напряжения в результате сокращения.Миотония Томсена - причины, симптомы, диагностика и лечение
Миотония Томсена — наследственно-семейное поражение поперечно-полосатой мускулатуры, проявляющееся пролонгированным расслаблением мышц после их сокращения. Кроме типичного миотонического феномена, заболевание характеризуется гипертрофическими изменениями пораженных мышц, манифестацией с поражения кистей, частой вовлеченностью лицевой мускулатуры. Миотония Томсена диагностируется на основании анамнестических данных, результатов неврологического осмотра, ЭМГ и ЭНГ, молекулярно-генетического исследования. Лечение сводится к назначению препаратов, уменьшающих тонические реакции мышц и нормализующих ионное состояние миофибрилл.
Общие сведения
Миотония Томсена входит в группу наследственных миотоний, включающую также миотонию Россолимо-Штейнерта-Куршмана, врожденную парамиотонию Эйленбурга, миотонию Беккера и еще ряд заболеваний. Болезнь Томсена приобрела свое название в соответствии с фамилией ученого, подробно описавшего ее симптомы и порядок наследования на примере 4-х поколений своей семьи. За 2 года до Томсена (в 1874 г.) первое описание заболевания дал Лейден. Поэтому в современной неврологии употребляется также название болезнь Лейдена-Томсена.
У больных с данным видом миотонии прослеживается доминантный тип наследования патологического аутосомного гена. Частота встречаемости заболевания составляет по различным оценкам от 3 до 7 человек на 1 млн. населения.
Миотония Томсена
Этиология и патогенез миотонии Томсена
Миотония Томсена относится к наследственным каналопатиям. Как и миотония Беккера, заболевание связано с дефектом 7-й хромосомы, а именно гена CLCN1, детерминирующего синтез белка хлорных ионных каналов миофибрилл скелетной мускулатуры. Следствием нарушения синтеза этого специфического белка является уменьшенное прохождение ионов хлора внутрь миофибриллы и их скопление на поверхности мембраны мышечного волокна (сарколеммы). Возникающая в результате биоэлектрическая нестабильность сарколеммы обуславливает ее чрезмерную возбудимость. При этом периферический неврон функционирует без отклонений, но на обычный нервный импульс мембрана миофибриллы реагирует повышенным возбуждением, которое препятствует нормальному расслаблению мышечного волокна после его сокращения. Причем, чем быстрее происходит сокращение миофибриллы, тем более затрудненно ее расслабление. Однако после серии сокращений реализуются компенсаторные механизмы, ионные каналы начинают усиленно функционировать и ситуация нормализуется.
Морфологические изменения мышечной ткани неспецифичны для миотонии Томсена и являются типичными для большинства миотоний. Отмечается централизация ядер сарколеммы, увеличение площади сечения миофибрилл, свидетельствующее о их гипертрофии. Электронная микроскопия определяет гипертрофию саркоплазматического ретикулума, увеличение размеров митохондрий и изменение их формы, утолщение телофрагмы.
Симптомы миотонии Томсена
В большинстве случаев манифестация болезни приходится на ранний детский возраст. Ведущим в клинике заболевания является миотонический симптомокомплекс, так называемый миотонический феномен. Он характеризуется пролонгированной мышечной релаксацией после совершения быстрого движения. Клинически это проявляется невозможностью быстро произвести следующее движение из-за тонического спазма мышц, участвовавших в предыдущем двигательном акте. Примечательно, что после серии активных движений скорость мышечного расслабления нормализуется и пациент может двигаться практически как здоровый человек. Перерыв в работе мышц приводит к возобновлению проблем с мышечной релаксацией в начале новой фазы двигательной активности.
Патогномоничным для болезни Томсена является ее манифестация с патологических изменений в кистях. Затем миотонические проявления распространяются на мускулатуру ног, мимическую и жевательную группы мышц. В результате тонические спазмы наблюдаются при резком начале ходьбы, зажмуривании глаз, смыкании зубов и т. п. движениях. При попытке быстрой ходьбы пациенты утрачивают равновесие и могут упасть из-за развития общей скованности. Эмоциональные переживания и пребывание в холоде усиливают миотонические реакции.
Своеобразен внешний вид пациентов с миотонией Томсена. Вследствие диффузной мышечной гипертрофии они зачастую имеют телосложение атлетов. Их мышцы отличаются чрезмерно плотной консистенцией, но имеют сниженную силу. Мышечная гипертрофия является отличительным симптомом заболевания, позволяющим дифференцировать его от миотонической дистрофии Россолимо-Штейнерта-Куршмана, сопровождающейся атрофиями мышц.
Диагностика миотонии Томсена
Поскольку миотония Томсена обычно манифестирует в раннем возрасте, родители заболевшего ребенка обращаются прежде всего к педиатру, который и направляет их на консультацию детского невролога. Для выявления миотонического симптомокомплекса невролог просит обследуемого несколько раз быстро сжать и разжать кулак. Замедленное разжимание пальцев в начале тестирования и постепенная нормализация движений при их повторении свидетельствуют в пользу миотонии. О наличие миотонического феномена говорит образование «валика» локального мышечного сокращения в ответ на постукивание неврологическим молоточком по мышце. В неврологическом статусе выявляется снижение мышечной силы и миотонический характер сухожильных рефлексов. Возможна миотоническая реакция зрачков на световое раздражение.
Основополагающими в диагностике миотонии Томсена являются электрофизиологические исследования: электромиография и электронейрография. Первая выявляет специфичные для миотонии повторяющиеся высокочастотные разряды, а вторая дает возможность полностью исключить поражение анимальной нервной системы.
Биопсия мышц позволяет изучить морфологические изменения мышечной ткани. Тем не менее, выявляемая при болезни Томсена гипертрофия миофибрилл и централизация ядер их сарколеммы наблюдается и при других типах миотоний. Для постановки точного диагноза с верификацией вида миотонии необходимо проведение молекулярно-генетического анализа, позволяющего определить дефект гена CLCN1.
Лечение миотонии Томсена
Современной медициной еще не разработана радикальная терапия генных заболеваний. Поэтому основная задача лечения миотонии Томсена сводится к уменьшению ее проявлений и замедлению прогрессирования симптомов. Для снижения тонической спастичности используются такие препараты, как дифенин, фенитоин, карбамазепин. Для поддержания ионного равновесия мембран миофибрилл применяют препараты кальция в виде медикаментозной терапии, электрофореза, гальванизации воротниковой зоны. С этой же целью рекомендованы мероприятия по уменьшению содержания калия в организме: диета с малым содержанием калия, мочегонная терапия. Из мочегонных средств предпочтительно назначение ацетазоламида, поскольку, кроме усиленного выведения калия с мочой, он также увеличивает проницаемость сарколеммы.
Особое значение в комплексном лечении миотонии Томсена имеют массаж и ЛФК. Они должны проводиться с особой осторожностью, чтобы улучшить обменные процессы мышечной ткани и одновременно уменьшить ее наклонность к тоническому спазмированию.
Прогноз и профилактика
Миотония Томсена сохраняется в течение всей жизни. Доброкачественное течение и медленное прогрессирование болезни делает ее прогноз относительно благоприятным. Пациенты, как правило, сохраняют трудоспособность. Однако, их профессия не должна быть связана с необходимостью совершать быстрые движения. Такие люди не могут работать на конвейере, быть водителями, пожарными, полицейскими и т. п.
К мерам первичной профилактики болезни Томсена можно отнести консультирование генетиком семейной пары, планирующей беременность и имеющей случаи заболевания среди родственников. Профилактикой миотонических приступов служит исключение резкой двигательной активности, переохлаждения, физических перегрузок и эмоционального волнения.
Миотонический синдром - Спазмы(судороги)
Автор На чтение 9 мин. Просмотров 2 Опубликовано
Миотонический синдром причины
Миотонический синдром может быть врождённым заболеванием и проявиться уже в первые месяцы жизни человека. К основным причинам его возникновения врачи относят поражения ядер подкорки головного мозга, патологию мозжечка и сбои в работе вегетативных нервных центров. Если же проявления синдрома на обусловлены генными причинами, можно сделать вывод что его появление обусловлено детскими патологиями, такими, как рахит, сбои течения процесса обмена веществ, травмы, полученные при родах, нарушения внутриутробного развития плода.
Этиология и патогенез миотонии
Среди двух типов дистрофической миотонии тип 1, ген которого картирован в локусе 19q13, встречается наиболее часто (98%). Как и все типы дистрофической миотонии он передается по наследству по аутосомно-доминантному типу. Основной этиологический фактор дистрофической миотонии 1 типа — увеличением количества тринуклеотидных повторов CTG (до нескольких тысяч). Миотонин-протеинкиназа, кодируемая геном DMPK, присутствует не только в скелетных, но и миокарде, а также ЦНС. Этим и объясняются основные клинические проявления дистрофической миотонии.
Врожденная парамиотония Эйленбурга, связанная с патологией натриевых каналов, передается по аутосомно-доминантному типу. Ген SCN4A картирован в локусе 17q23.1-q25.3. Парамиотонические проявления развиваются в связи с повышенной возбудимостью мембраны мышечных волокон и нарушением функционирования сократительных элементов мышцы.
В отличие от большинства нейромиотоний, являющихся приобретенными заболеваниями, идиопатическая нейромиотония (синдром Исаакса) передается по наследству и встречается наиболее часто. Эффективность введения кураре для купирования мышечных спазмов говорит о неврогенной природе нейромиотонии, однако окончательно патогенез заболевания неясен и сегодня. С выявлением повышенного титра антител к потенциал-зависимым калиевым каналам нейромиотонию стали считать аутоиммунным заболеванием. Об аутоиммунном характере заболевания говорит также наблюдаемая в ряде случаев эффективность плазмафереза.
Миотонический синдром симптомы
Основным симптомом болезненного состояния выступает спазм мышц. После того, как мышца претерпела резкое сокращение её не получается расслабить сразу же, для этого необходимо некоторое количество повторяющихся движений. Непродолжительный отдых также способствует острым проявлениям тонических напряжений. Самые первые движения больной проделывает с величайшими затруднениями, для полного расслабления мышц необходимо не менее полминуты.
Симптомы миотонического синдрома, проявляющиеся на холоде, могут полностью исчезнуть при попадании в тепло. Продолжительность мышечного сокращения варьируется от двух трёх минут до нескольких часов. После этого на протяжении нескольких суток может присутствовать слабость в мышцах, после прекращения воздействия теплоты на организм.
Для миотонической дистрофии характерна атрофия развития мышечных групп области лица, шеи, а также для описываемого синдрома возможны проявления спазмов жевательных мышц, дистальных отделов конечностей.
Рефлексы миотонического синдрома нередко проявляются в передней лестничной мышце, которая имеет локализацию в шейном отделе позвоночника, а также в грушевидной мышце.
Миотонический синдром относится к числу болезней мышечной и нервной систем. Его проявления заключаются в понижении мышечного тонуса, что означает сложности с расслаблением мышц различных групп после того, как оно испытали сильное напряжение. Сокращение мышцы можно наблюдать после того, как будет сначала нанесен лёгкий удар по мышце при помощи пластмассового молотка или пальцем. Сокращение может быть весьма длительным и неприятным. В какое либо иное время мышцы ребёнка могут пребывать в пониженном тонусе.
Вялая осанка ребёнка может являться следствием его мышечной слабости, которая сопутствует проявлению миотонического синдрома. Дети с подобными проявлениями начинают двигаться и ходить значительно позже своих сверстников, поскольку им нелегко ровно держать спину и они испытывают боли в суставах, вызванные нарушениями их функционирования. Вследствие нарушения мышечного тонуса и говорить такие дети начинают значительно позже. Их речь при этом невнятна и невыразительна.
Для чёткого определения проявления миотонического синдрома у взрослого человека необходимо проведение врачом физиакального обследования посредством разных тестирований, одним из которых можно назвать сжатие и разжатие мячика. Очень важно при этом точно установить, является ли миотонический синдром прогрессирующей болезнью, или же имеют место проявления атрофической миотонии либо какой-нибудь иной болезни.
При миотоническом синдроме, в целях облегчения ситуации для больного и назначения правильного курса лечения рекомендуется проведение исследований в лаборатории. Электромиография способна зарегистрировать параметры электрической активности мышц скелета, кроме того требуется проведение биопсии мышц, генетического анализа, исследований крови.
Лечение миотонии
Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии — снижение выраженности миотонических проявлений. Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции.
Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин, а для снижения уровня калия — диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека, преднизолон, циклофосфамид.
От того, насколько быстро был обнаружен миотонический синдром, зависит скорость, с которой он будет устранён и излечен. Некоторые врачи разделяют мнение относительно неэффективности терапии миотонического синдрома при помощи медицинских препаратов, однако регулярные занятия физкультурой способны оказать надлежащий эффект. Нагрузка при этом должна быть распределена специалистом.
Препараты
В определённых случаях для снятия мышечных спазмов можно применять препарат Фенитоин. Он представляет собой мышечный релаксант, действие которого относится к числу противосудорожных. Расчёт необходимой дозировки производится в соответствии с весом больного человека и его возраста. Для взрослого человека суточная дозировка недолжна превышать 500 мг, для ребёнка 300 мг в течение суток. Кроме того при миотоническом синдроме можно рекомендовать следующие препараты:
- Актовегин. Повышает энергетический потенциал клеток организма человека, ускоряет течение метаболизма. Имеется в виде таблеток, также можно вводить внутривенно;
- Пантогам. Также вызывает активизацию процесса метаболизма в клетках организма, действие его противосудорожное. Устраняет возбудимость и неврозы. С его помощью проводят профилактику и лечение задержек умственного развития у детей;
- Элькар. Представлен в виде капель, оказывающих воздействие на процессы активности мозга и ускоряющих метаболизм. Рекомендован также при слабости мышц и дистрофии.
Массаж
Массаж при описываемом синдроме эффективен, если он проводится достаточно интенсивно. Сперва необходимо тщательно размять массируемую область, после чего тщательно и интенсивно помассировать на протяжении 15-20 минут. В конечном счёте решающее значение имеет интенсивность проведенных операций. Рекомендуется проводить массаж ежедневно на протяжении недели для существенного облегчения состояния больного.
Электрофорез
Проведение электрофореза относится к разряду процедур физиотерапии. Его суть заключается в том, что применяемое при лечении вещество аккуратно наносится на электродные прокладки и посредством воздействия магнитного поля вводится в организм человека. Именно в месте введения средства оказывается наибольшее воздействие на процессы, приводящие к миотоническому синдрому. Электрический разряд известен своим гуморальным и нервно-рефлекторным воздействием.
Гимнастика
Выполнение гимнастических упражнений обеспечивает должный уровень двигательной активности всех мышечных групп организма. Соответствующий подбор необходимых упражнений рекомендуется проводить, консультируясь с грамотным специалистом в этой области, однако следует заметить, что интенсивность упражнений следует увеличивать постепенно, во избежание перегрузки подвергшихся заболеванию участков.
Клиническая картина миотонии
Симптом «кулака» — основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов — миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика», возникающие при введении и перемещении игольчатого электрода.
Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. Дистрофическая миотония — мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). Для парамиотонии типична т. н. «холодовая миотония» — возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Диагностика миотонии
Дебют дистрофической миотонии приходится, как правило, на юношеский либо взрослый возраст, степень прогрессирования заболевания зависит от генетического дефекта, поэтому тщательный сбор семейного анамнеза имеет большое значение в диагностировании дистрофической миотонии. Миотония Беккера дебютирует в 5-12 лет и характеризуется медленным течением, а миотония Томсена может дебютировать как в детском, так и в зрелом возрасте и протекает, как правило, тяжело и с осложнениями. Физикальное обследование при дистрофической миотонии выявляет атрофию мышц и снижение их силы. Для дистрофической миотонии типа 1 характерна мышечная слабость в дистальных отделах конечностей, для дистрофической миотонии типа 2 — в проксимальных.
С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови.
Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды — патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Для врожденной миотонии типично сохранение параметров ДЕ в пределах нормы, для дистрофической миотонии — сочетание невропатических и миопатических черт. Для диагностики парамиотонии проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества CTG-повторов в гене DMPK.
Дифференциальный диагноз
Как правило, дифференцировать врожденную миотонию от дистрофической миотонии неврологу удается по клиническим признакам. Однако в ряде случаев врожденной миотонии определяют легкую слабость дистальных мышц рук и слабую активность при ЭМГ — признаки, типичные для дистрофической миотонии. Постоянная мышечная активность — клинический признак нейромиотонии — входит в состав синдрома “ригидного человека” (stiff-man syndrome), однако в отличие от нейромиотонии мышечная активность при синдроме “ригидного человека” снижается после введения диазепама, а также во время сна.
Миотонический синдром у взрослых симптомы. Миотонический синдром причины, симптомы, методы лечения и профилактики
Наследственные миотонические синдромы (НМС) - группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами [1 ] дистрофических (ДМ) и [2 ] недистрофических миотоний (НДМ).
о клинической и ЭМГ феноменологии миотонии вы можете прочитать в статье «Клинико-диагностические критерии миотонии» Н. А. Шнайдер; ГБОУ ВПО Красноярский государственный медицинский университет имени проф. В. Ф. Войно-Ясенецкого Министерства здравоохранения РФ, кафедра медицинской генетики и клинической нейрофизиологии ИПО (журнал «Сибирское медицинское обозрение» №3, 2016) [читать ]
Недистрофические миотонии . Самая распространенная форма НДМ - врожденная миотония (ВМ), с распространенностью от 1 до 9,4 на 100 тыс. населения, что зависит от страны и этнической принадлежности. Происхождение ВМ обусловлено мутациями в гене хлорного канала CLCN1 (локус 7q35) в поперечно-полосатых мышцах (нарушается проводимость ионов Cl- в клетку, что ведет к повышенной возбудимости мышечной мембраны и мышечной ригидности). ВМ клинически проявляются:
[1 ] генерализованными миотоническими феноменами;
[2 ] гипертрофией скелетной мускулатуры;
[3 ] транзиторной слабостью;
[4 ] дебютом в раннем возрасте;
[5 ] стационарным течением и благоприятным прогнозом.
На сегодня выделяют 2 формы ВМ. Первой описана миотония Томсена (МТ) с аутосомно-доминантным типом наследования и лишь 100 лет спустя - миотония Беккера (МБ) с аутосомно-рецессивным типом наследования. Молекулярно-генетические данные показали, что ранее считавшаяся редкой ВМ Беккера встречается даже чаще, чем ВМ Томсена. При дебюте в раннем возрасте пациенты с МТ и МБ имеют, как было указано выше, гипертрофию скелетных мышц («атлетическое сложение», более характерное для ВМ Томсена), иногда легкую миалгию, слабость, задержку мышечного расслабления, а также относительно стационарное течение и благоприятный прогноз. То есть больные с МТ и МБ имеют однотипные клинические проявления, различаясь лишь степенью их выраженности (ВМ Беккера считают несколько более тяжелой, но не в каждом случае).
Дистрофические миотонии (или ДМ, или сongenital myotonic dystrophy, myotonic dystrophy - DM) является мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Это наиболее распространенное заболевание из класса миотоний. Клиническая картина дистрофической миотонии складывается из 3 синдромов:
[1 ] миотонический синдром;
[2 ] дистрофический синдром;
[3 ] синдром вегетативно-трофических нарушений.
Таким образом, ключевая особенность ДМ - сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). МД характеризуется варьиру ющим началом заболевания: от пренатального периода до 50 - 60 лет. Различают четыре формы по возрастному «пику» начала заболевания: [1 ] врожденная (син.: конгенитальная ДМ или congenital myotonic dystrophy ; клиническая симптоматика развивается сразу после рождения), [2 ] юношеская (син.: ювенильная МД или juvenile DM; с дебютом от 1 года до подросткового возраста), [3 ] классическая (син.: ДМ взрослых или adult DM; - с дебютом у индивидуумов старше 20, но моложе 40 лет) и [4 ] минимальная (син.: МД с поздним дебютом или DM with late onset; у индивидуумов старше 40 лет [как правило, до 60 лет] и с более легким течением). Это объясняется различиями в числе тринуклеотидных повто ров (CTG) в локусе гена МД, кодирующего синтез миотонин-протеинкиназы. Кроме различия в возрасте начала проявления болезни имеются достаточно различные клинические признаки разных подтипов этого заболевания.
До 1994 года ДМ считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: DM1 (мутация 19q13.3), DM2 (мутация 3q21) и DM3 (мутация 15q21-q24). Существуют отдельные исследования, подтверждающие наличие четвертого подтипа DM и др. (DM4, DMX). Наиболее часто встречается (среди всех миотоний, как среди ДМ, так и среди НДМ) DM1 - ДМ Россолимо-Куршмана-Штейнерта-Баттена (с частотой 13,5 на 100000 живых новорожденных; распространенность в больших популяциях около 1: 8000). Распространенность DM2 (болезнь Thornton-Griggs-Moxley) и DM3 в настоящее время недостаточно изучена.
читайте также пост: Креатинкиназа (справочник невролога) (на сайт)
В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления ДМ вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с ДМ могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.
подробнее о DM1, DM2 и DM3 читайте [1 ] в статье «Клинико-генетическая гетерогенность дистрофической миотонии» Н.А. Шнайдер, Е.А. Козулина, Д.В. Дмитренко; Кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования ГОУ ВПО «Красноярская гос. мед. академия Федерального агентства по здраво-охранению и социальному развитию», Россия (Международный неврологический журнал, №3, 2007) [читать ] (или [читать ]) и [2 ] в статье «Миотоническая дистрофия. Современное представление и собственное наблюдение» Т.И. Стеценко, Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев, Украина (журнал «Современ-ная педиатрия» №1, 2014) [читать ]
Обратите внимание ! Для НДМ не характерно развитие парезов конечностей и мышечных атрофий в отличие от больных с ДМ1, у которых вялые дистальные парезы рук и ног служат главной причиной инвалидизации. Тем не менее, у больных с НДМ (форма Томсена, Беккера) специфическими симптомами могут выступать исходная неловкость и мышечная слабость в кистях, обусловленная типичными миотоническими задержками и исчезающая после нескольких повторных произвольных сокращений мышц. Симптом получил наименование «транзиторная слабость», а уменьшение выраженности миотонии при повторных мышечных сокращениях - «феномен врабатывания». Транзиторная слабость отсутствует у больных с ДМ1, а слабость в мышцах кисти остается неизменной даже после уменьшения миотонических проявлений. Наличие транзиторной слабости у больных с НДМ связывали с дебютом ДМ1, что затрудняло дифференциальную диагностику, приводило к неверным нозологическим трактовкам и неадекватным генетическим прогнозам в семьях пробандов. Все это послужило поводом к необходимости выработки объективных критериев наличия или отсутствия транзиторной слабости. Проведение теста с ритмической стимуляцией (РС) с частотой 10 - 60 Гц у больных с НМС выявило декремент М-ответа с восстановлением его амплитуды после тетанизации. Кроме того, при проведении РС у больных с НМС на примере мышц кисти, иннервируемых локтевым нервом, выявляется зависимость между величиной декремента амплитуды М-ответов и выраженностью транзиторной слабости. Выявленный декремент М-ответа при РС в генотипированной группе больных с НДМ объясняется нарушением функции хлорных каналов. [!!! ] Таким образом, декремент амплитуды М-ответов может быть информативным показателем в алгоритме диагностического поиска мутаций в гене хлорного канала у па
Миотонический синдром левой короткой мышцы головы симптомы. Как лечить миотонический синдром у грудных детей? Что можете сделать вы
Любой родитель желает только одного – чтобы малыш родился здоровым и не имел никаких серьезных отклонений в развитии или заболеваний. К огромному сожалению, даже сильного желания не достаточно для того, чтобы избежать возможных проблем – каждая болезнь имеет свою статистику, и порой цифры крайне неутешительны. Узнать о том, что у малыша проблема со здоровьем еще во время его внутриутробного развития порой просто невозможно, многие патологии обнаруживают себя уже после появления на свет. Не исключение и миотонический синдром, входящий в группу заболеваний нервно-мышечного типа.
Некоторые специалисты не классифицируют данный синдром как болезнь, а говорят о нем как о симптоматическом проявлении мышечных нарушений. У пациентов с таким диагнозом нарушена способность мышц к быстрой релаксации после очередного напряжения, то есть процесс расслабления проходит намного дольше, чем у здорового человека. При наличии такой патологии движения ребенка, особенно первые, даются огромными усилиями. Как распознать наличие болезни у малыша и как помочь ему? Это и попробуем выяснить.
Разновидности и причины появления синдрома
При миотоническом синдроме мышцы не способны быстро восстанавливаться
На сегодняшний день группу миотонических заболеваний описывают с разных точек зрения. Так, некоторые включают в нее исключительно врожденные формы миотонии, а некоторые – любые нарушения, касающиеся способности мышц к расслаблению, даже те, что носят временный характер. Классическая форма миотонического синдрома – врожденная. Точные механизмов ее появления у детей не установлено, но выделены предполагаемые причины ее возникновения:
- наследственный фактор, то есть особенности мышечного тонуса передаются ребенку от родителей или кровных родственников;
- нарушения обмена веществ у будущей мамы;
- прием определенных медикаментозных препаратов во время беременности.
Приобретенная форма может становиться следствием малоактивного образа жизни ребенка, перенесенного в раннем возрасте рахита, нарушений нормального обмена веществ в организме. Комплекс мышечных нарушений в рамках миотонического синдрома свойственен в разных своих формах следующим заболеваниям:
- синдром ригидного человека;
- синдром Шварца-Джампела;
- столбняк;
- злокачественная форма гипертермии;
- нейролептический синдром;
- энцефаломиелит.
Характерные для миотонии симптомы появляются также у тех людей, которых укусил ядовитый паук Черная вдова.
Классическая форма миотоничесокго синдрома – это миотония Томпсона. Заболевание носит наследственный характер и передается по аутосомно-доминантному признаку. Начаться оно может в любом возрасте, но зачастую это происходит либо в детстве, либо в период юношества. При наличии патологии в такой форме, наследование в большинстве случаев случается в каждом поколении без исключений, а для передачи болезни детям достаточно наличия проблемы хотя бы у одного родителя.
Миотонический синдром передается по наследству
Вторая форма заболевания, которая встречается реже, – миотония Беккера, передающаяся по аутосомно-рецессивному типу. В такой ситуации болезнь обычно случается лишь в одном поколении на фоне отсутствия подобных проблем в клинической картине семьи . Если оба родителя выступают носителями измененного гена, то с вероятностью в 25% ребенок родится с таким диагнозом. Если же только один из родителей выступает носителем, то есть вероятность того, что чадо также будет носителем, но симптоматических проявлений это давать не будет.
Симптомы проблемы
В основе постановки диагноза лежит специфическое проявление болезни, а именно расстройство двигательного акта. После того, как мышца сократилась, не происходит естественного расслабления, напротив, возникает тоническое напряжение. Сложнее всего даются первые шаги после какого-то периода спокойствия, уже через несколько циклов сокращения-расслабления процесс проходит немного легче. Фаза расслабления мышцы обычно затягивается до 30-ти секунд.
Впервые наличие врожденной проблемы родители могут заметить не сразу. Ярче всего симптомы проявляются во время активности – подъем по лестнице, быстрая ходьба или прочие движения, которые можно отнести к числу сложно скоординированных. Дети с врожденной миотонией начинают позже ходить и говорить, плохо держат спинку.
Признаки на фото
Даже на слабые внешние раздражители мышцы ребенка с таким диагнозом реагируют ярко, возбудимо. Так, также легкий удар игрушкой или пальцами по определенной группе мышц может привести к сильному спазму – на месте удара возникает напряженный мышечный валик, который очень долго расслабляется.
Заболевание специфическое и, как ни странно, сопровождается мышечной слабостью. Больные дети обычно имеют слабый мышечный корсет спины, что приводит к формированию неправильной осанки, слабые мышцы брюшного пресса, что провоцирует проблемы желудочно-кишечного тракта. Слишком слабые мышцы в зоне лица становятся причинами многих проблем, в том числе нарушений зрения (близорукости) и плохого развития речевой функции.
Проще всего заметить так называемую миотоническую атаку. Например, ребенок пытается встать, но почему-то замирает на какое-то время или вовсе падает, но после нескольких повторных усилий ему все удается подняться, зачастую с помощью какой-либо опоры. Резкие движения сопровождаются более сильными спазмами – когда малыш хочет быстро перейти на бег, то он как бы обездвиживается и часто просто падает словно парализованный. В большинстве случаев, когда ребенок немного «расходится», движения даются ему нормально.
Когда болезнь присутствует уже во взрослом возрасте, то можно отметить ее наличие визуально – подросток выглядит накачанным в тех группах мышц, которые подвержены постоянному напряжению. Это могут быть как мышцы спины или ног, что выглядит эстетично, так и мышцы лица и шеи, что уже не имеет столь привлекательного вида.
Как вылечить миотонический синдром у ребенка
Современная медицина не может на данный момент предложить способа, который бы помог полностью избавиться от проблемы. Но опускать руки не стоит – благодаря симптоматическому лечению ребенок сможет вести нормальный образ жизни. При наличии интенсивных атак назначается курс медикаментозной терапии, который помогает минимизировать их. Если же симптомы не имеют ярких проявлений, то лечение ограничивается следующими мероприятиями:
Для улучшения эффекта врачом могут быть назначены физиотерапевтические процедуры (например, иглорефлексотерапия или электрофорез) и рекомендованы занятия по плаванию.
Диагноз «детский миотонический синдром» - это не приговор, комплексный подход и следование рекомендациям специалистов позволят ребенку нормально развиваться и вести активную жизнь в самостоятельном будущем .
Видео о детском массаже
Виды миотонического синдрома
Миотоническим синдромом называется феномен расслабления мышц после их сильного напряжения. Существенные усилия при высоком уровне двигательной активности провоцируют дальнейшее развития указанного синдрома. Расслабляющая фаза после того, как произведено чувствительное усилие растягивается по времени на полминуты. Миотонический синдром предполагает существенные затруднения двигательной активности больного после совершения первых же движений, в дальнейшем ему становится легче. Движения его приобретают дополнительную лёгкость и по прошествии определённого п
Миотонический синдром у детей — причины, симптомы и лечение
Здоровые, улыбающиеся дети — самая большая радость для родителей. Но не всегда бывает так, что у заботливых и внимательных пап и мам растут не болеющие малыши.
В педиатрии есть такие состояния, о которых родители просто не знают и не догадываются. Поэтому, когда врач поставит диагноз ребенку «миотонический синдром», то это зачастую является ударом для его родственников. Что же это за состояние, каковы его симптомы, угрозы, и чем лечить болезнь?
Симптомы миотонического синдрома у детей
После того, как родители услышат подобный диагноз, паниковать не нужно, да и времени на это нет. Следует оперативно приниматься за лечение и сделать все, чтобы вылечить ребенка, устранить возникающие угрозы.
Миотонический синдром — заболевание нервно-мышечного характера. Оно характеризуется снижением общего тонуса мышц. Когда они отвечают сокращением на внешние раздражители, то затем с большим трудом расслабляются. Такая мышечная слабость всегда формирует у мальчика или девочки вялую осанку. Дети значительно позже своих сверстников начинают ходить, им тяжело держать спину ровной. Нередко у таких деток наблюдаются нарушения работы суставов. Следует отметить, что пониженный тонус характерен всем мышцам, в том числе и мышцам лица, языка, по этой причине дети и говорить начинают гораздо позже. А если речь развивается вовремя, то с большими нарушениями, невыразительностью.
Слабый мышечный каркас таких детей в зоне пресса приводит также к нарушениям работы желчно-выводящих путей, к запорам. Иногда низкий тонус мышц провоцирует энурез, а слабость глазных мышц — близорукость. Дети с миотоническим синдромом устают быстрее других малышей, они утомляются даже от небольших умственных и физических нагрузок. Это значит, что учиться им будет трудно. Ведь практически регулярно таких деток мучают головные боли. С детства у них развивается сколиоз и остеохондроз, может деформироваться позвоночник, что, конечно же, влияет и на психическое состояние детей.
Читайте такжеВсе эти признаки миотонического синдрома заметны не сразу после рождения, и, возможно, родителям самим даже трудно вначале диагностировать какие-либо отклонения. Симптомы миотонического синдрома выявляет педиатр. Поэтому следует регулярно посещать с ребенком детскую консультацию, ходить на плановые медицинские осмотры, чтобы вовремя диагностировать возможные проблемы развития малыша.
Причины возникновения
Врачи не зря говорят, что беременность надо планировать. В организме здоровой мамы, которая серьезно относиться к появлению на свет своего первенца, формируются здоровые половые клетки. Прием фолиевой кислоты еще до наступления беременности обезопасит родителей от появления на свет нездорового ребенка, предотвратит пороки его внутриутробного развития.
Причиной митонического синдрома у детей бывают и генетические факторы, а именно особенности тонуса мышц, которые передаются по наследству. Нередко возникновение вышеуказанной болезни у детей — это последствия рахита и нервно-мышечных заболеваний, нарушения обмена веществ и гиподинамия самой мамы в период вынашивания ребенка.
Лечение миотонического синдрома у детей
Прежде всего, нужно организовать здоровый и активный образ жизни малыша после его появления на свет. Это означает кормление грудью и прогулки на свежем воздухе в любое время года, массажи и ежедневную гимнастику, зарядку по утрам. Такие процедуры станут хорошими профилактическими мероприятиями, чтобы избежать дальнейшего прогресса слабости мышц. Но не всегда этих мер бывает достаточно. Речь идет о случаях, когда причиной миотонического синдрома являются генетические факторы. Тогда назначается квалифицированное лечение, к которому привлекаются логопеды и ортопеды, терапевты и окулисты. Для успешного устранения миотонического синдрома у малыша надо прибегнуть к курсу массажа и обязательно пройти полный курс физиотерапевтических процедур. Иногда назначается и медикаментозный курс лечения. Доктора приписывают препараты, улучшающие обменные процессы и повышающие нервно-психическую проводимость.
Можно также лечить детей с миотоническим синдромом в специальных медицинских центрах, где разработаны программы коррекции здоровья. Они включают физиотерапию и занятия с логопедом, психологом, иглорефлексотерапию.
Миотонический синдром. Симптомы и лечение. Миотония – описание и классификация синдромов, причины и лечение заболевания Шейный миотонический синдром
И сбоях в функционировании их волокон. Патологический процесс в основном наблюдается в детском возрасте, однако также отмечаются случаи во взрослом. При появлении неприятной симптоматики следует обратиться к специалисту, чтобы в кратчайшее время начать лечение.
В медицинской практике существуют такие виды рассматриваемого заболевания:
- Миотония Томсона. Является аутосомно-доминантным видом патологии, передающейся от одного из родителей. Первоначальная симптоматика нарушений проявляется в младенчестве, однако отмечаются ситуации, когда болезнь напоминает о себе лишь в 7-12 лет.
- Миотония Беккера. Болезнь является аутосомно-рецессивным видом миотонии и отмечается значительно чаще, чем вышеуказанный тип. Формирование симптомов у девочек отмечается раньше (5-12 лет). У мальчиков проявляются в 16-18 лет.
- Синдром Шварца-Джампела. Наблюдается уже по достижении 12 месяцев. У малыша отмечаются аномалии в скелете, черепе врожденного характера, умственными нарушениями. Синдром способен передаваться генетическим путем лишь от двух родителей.
- Дистрофическая миотония (заболевание Куршмана-Штейнерта). Включает в себя нервно-мышечные нарушения и патологические процессы в глазах, вегетативной и эндокринной системах.
- Врожденная парамиотония. Наблюдается нечасто и передается по аутосомно-доминантному типу. Совмещает симптоматику эпизодического паралича и миотонии.
Причины
Этиологические и патогенетические факторы миотонии полностью не установлены. Специалисты склонны выделять 2 формы патологического процесса:
- Врожденная. Спровоцировать болезнь способны сбои генетического характера. Синдром может передаваться наследственным путем от родителей. Чтобы заболевание проявилось клинически, требуется получить от больных родителей по 1 измененной генетической копии. В такой ситуации мутировавший ген будет подавлять его нормальную копию, вследствие чего развивается патологический процесс. Когда ребенок унаследовал измененный ген лишь от одного родителя, он станет носителем, но болезнь не проявится.
- Патологическая. Вызвать формирование патологического процесса способны нервно-мышечные и эндокринные болезни, травматизм во время родов, энцефалопатия младенцев, обусловленная кислородным голоданием, сбои в обмене веществ, пассивный образ жизни при беременности на протяжении всего периода. Приобретенная миотония действенно лечится, в отличие от генетической формы синдрома.
Механизм формирования заболевания одинаков у каждого пациента. Ослабленные мышцы под влиянием некоторых факторов существенно тонизируются и парализуются. Может наступить миотоническая атака. Она формируется во время попыток больным совершить движение, требующее участия поврежденных мышц. К провоцирующим факторам относят переохлаждение, стрессовые ситуации, эмоциональное потрясение, шум, продолжительное нахождение в одной позе.
Симптомы
Клиническая симптоматика патологического процесса зависит от нахождения воспалительного очага. В процессе нарушения мышечной активности в ногах, руках, плечах, шейном отделе, лице у пациентов проявляются сложности при передвижении, удержании осанки, во время диалога, мимики, трапезы. К общим симптомам относят:
- слабость в мышцах;
- быструю утомляемость;
- апатичное состояние;
- сбои в пищеварительном тракте, запоры и колики в кишечнике хронического характера;
- энурез;
- цефалгию невыясненной этиологии;
- , сгорбленность;
- миопию;
- расстройства речи;
- сбои в координации, утрату устойчивости;
- неуверенную походку во время крутого подъема и резкого спуска;
- ухудшение интеллектуальных способностей.
При проявлении неприятных признаков, следует в кратчайший период провести диагностику и подобрать соответствующее лечение.
Миотонический синдром у детей
Первоначальные проявления миотонического синдрома в детском возрасте. Больные дети позднее здоровых начинают удерживать голову, передвигаться и разговаривать. Данные действия они осуществляют со значительными усилиями и зачастую лишь при помощи взрослых.
Ребенок не в состоянии удерживать спину, прямо сидеть либо стоять. Он устает при наименьших усилиях. Также у него отсутствует желание играть, в частности в различные подвижные игры, отсутствуют увлечения, которые характерны для такого возраста. Со стороны пищеварения у него наблюдаются хронические запоры. Во время смеха, чихания, кашля и натуживания происходит недержание. Характерной чертой станет резкое формирование близорукости.
Если не обращать внимание на негативную симптоматику патологического процесса, возможно причинить существенный урон здоровью ребенка. В результате этого произойдут необратимые последствия.
Миотонический синдром у взрослых
В молодом и зрелом возрасте проявляются симптомы дистрофического типа миотонии (заболевание Штейнера). Зачастую подобное сопряжено со смежными заболеваниями сердечно-сосудистой системы, нарушениями в менструальном цикле у женщин и половым бессилием у мужчин. Кроме сбоев неврологического характера, отмечается существенное понижение интеллектуальных способностей.
Миотонический синдром во взрослом возрасте способенпроявиться достаточно своеобразно. Лица мужского пола с мышечной гипертрофией имеют спортивный и подтянутый вид, однако по факту мышцы не имеют нужного тонуса. Миотоническое спазмирование ведет к изменениям в голосе, нарушениям в процессе глотания и дыхания (повреждение мышц). Первоначальные мышечные спазмы преимущественно повреждают нижние конечности. Пациент в состоянии надлежащим образом передвигаться по ровному покрытию, однако когда потребуется совершить подъем по ступеням, использовать другие мышцы, задача значительно усложнится.
У страдающих миотонией возможно заметить феномен приседания. Он не сможет сесть таким образом, чтобы задняя часть бедра касалась голени и в полной мере опиралась на ступню. Пациент не способен сохранять равновесие в подобной позиции и попросту падает. Несложное для здоровых людей приседание выполняется широко расставив ноги и сделав упор на носки (в полной мере сесть не удастся).
Миотонический синдром, который начался во взрослом возрасте, нуждается в той же терапии, что и у детей (купирование дискомфорта). Также необходимо установить фактор, который спровоцировал формирование патологического процесса. Таким образом будут достигнуты сразу 2 цели: исчезнут мучительные судороги и спазмирование, а также исключается появление других последствий, способных вызвать основной недуг.
Диагностика
Терапевтические и диагностические процедуры во время миотонии осуществляют неврологи, логопеды, ортопеды, педиатры и офтальмологи. К основным диагностическим мерам относят:
- Изучение анамнеза. Выясняются сведения о присутствии похожего заболевания у близких.
- Простукивание мышц с помощью перкуссионного молотка в целях выявления дефектов.
- Специфический тест в целях обнаружения миотонической реакции на раздражителей - нагрузка, холод, громкие звуки. Изначально у пациентов возникают миотонические разряды, а затем исчезают характерные феномены. Пациенту предлагается быстро разжать кисть, открыть глаза, подняться со стула. Чтобы выполнить подобные несложные действия больному понадобится время и некоторые усилия.
- Э
причины и лечение Миотония томсона лечение
Описание:
Врожденная миотония (болезнь Лейдена–Томсена) впервые описана Лейденом в 1874 г. Томсен в 1876 г. обратил внимание на наследственную природу болезни на примере своей семьи (дети и многие ро дственники – 20 члено в его семьи в 4 поколениях страдали миотонией).
Часто та 0,3–0,7 на 100 000 населения.
Причины врожденной миотонии:
Наследуется по аутосомно-доминантному типу. Пенетрантность более высокая у лиц мужского пола.
Патогенез:
Имеют значение нарушения проницаемости клеточной мембраны, изменение ионного и медиаторного обмена (нарушения функциональной взаимосвязи в звене кальций– тропонин–актомиозин), повышенная чувствительность ткани к ацетилхолину и калию.
Патоморфология. При световой микроскопии обнаруживается гипертрофия отдельных мышечных во ло ко н; гистохимически о пределяется уменьшение размеров II типа мышечных волокон; при электронной микроскопии выявляются умеренная гипертрофия саркоплазматической сети, изменение формы и увеличение размера митохондрий, расширение телофрагмы миофибриллярных волокон.
Симптомы врожденной миотонии:
Впервые симптомы заболевания проявляются преимущественно в возрасте 8–15 лет. Ведущими признаками служат миотонические спазмы – затруднения расслабления мышц после активного напряжения. Миотонические спазмы локализуются в раз-личных группах мышц, чаще в мышцах кисти, ног, жевательных мышцах и круговых мышцах глаза. Сильное сжатие пальцев кисти, длительное статическое напряжение ног, смыкание челюстей, зажмуривание глаз вызывают тонические спазмы. Фаза расслабления мышц задерживается на продолжительное время, и больные не в состоянии быстро разжать кисти, изменить положение ног, открыть рот, глаза. Повторные движения уменьшают миотонические спазмы. Повышение механической возбудимости мышц определяется с помощью специальных приемов: при ударе неврологическим молоточком по возвышению I пальца происходит приведение его к кисти (от нескольких секунд до минуты) – «симптом большого пальца», при ударе перкуссионным молоточком по языку на нем появляется ямка, перетяжка – «симптом языка». Внешний вид больных своеобразен. Вследствие диффузных гипертрофии различных мышц они напоминают профессиональных атлетов. При пальпации мышцы плотные, твердые, однако объективно мышечная сила снижена. Сухожильные рефлексы нормальны, в тяжелых случаях снижены.
Течение. Болезнь медленно прогрессирует. Трудоспособность сохраняется в течение длительного времени.
Диагностика:
Диагноз строится на основании генеалогического анализа (аутосомно-доминантный тип наследования), особенностей клинической картины (атлетический тип телосложения, диффузные гипертрофии мышц, миотонический синдром), данных глобальной электромиографии (миотоническая реакция).
Дифференцировать заболевание следует от других форм миотоний, иногда – от псевдогипертрофических форм прогрессирующих мышечных дистрофий.
Леечние врожденной миотонии:
Назначают дифенин (по 0,1–0,2 г 3 раза в день в течение 2–3 нед), диакарб (по 0,125 г 2 раза в день в течение 2–3 нед), препараты кальция (внутривенно 10 % раствор хлорида кальция по 10 мл или глюконат кальция внутримышечно). Предполагается, что дифенин оказывает тормозящее влияние на моно– и полисинаптическое проведение в ЦНС, а диакарб изменяет проницаемость мембран. Целесообразны физиотерапия в виде гальванического воротника и трусов с кальцием, лечебная гимнастика.
Миотония - это группа наследственных заболеваний, характерный признак которых - замедленная мышечная релаксация после сокращения (миотонического феномена). Явление миотонии сопровождается миотоническими разрядами, которые диагностируются в процессе иг