Орфанные заболевания что это такое перечень
Орфанные заболевания по приказу Минздрава и список лекарств от редких болезней
«Сиротские» или орфанные заболевания – это группа редких болезней, которые поражают небольшое количество людей, патологии проявляются при рождении или в детском возрасте. Всего описано 7000 таких заболеваний, в России в список орфанных болезней включено 214 нозологий. Для исследования, разработки методов лечения данных патологий требуется помощь государства. «Сиротские» заболевания негативно влияют на качество жизни человека, способны стать причиной смерти.
Статьи по темеЧто такое орфанные заболевания
В медицинской науке не существует единого определения редких заболеваний. В одних странах орфанные патологии выделяют в зависимости от количества людей, страдающих от этого недуга, в других – от доступности способов лечения. В Америке считается, что редкие болезни затрагивают 1 человека из 1500, в Японии – 1 из 2500. В Европейских государствах к редким заболеваниям относят только хронические, угрожающие жизни патологии. В Российской Федерации орфанными патологиями считают те, которые встречаются не чаще 10 случаев на 100 000 человек.


Откуда берутся
Орфанные болезни в основном возникают из-за генетических отклонений, могут передаваться от родителей. Патологии имеют хронический характер. Считается, что большинство патологий проявляется сразу после рождения, редко симптомы могут проявиться во взрослом возрасте. Встречаются токсические, инфекционные, аутоиммунные «сиротские» заболевания. Выделяют следующие причины развития данных заболеваний:
- наследственность;
- плохая экология;
- пониженный иммунитет;
- высокая радиация;
- вирусные инфекции у матери во время беременности, у детей в раннем возрасте.
У детей
Большинство больных «сиротскими» заболеваниями рождаются с патологиями. Главная причина появления патологии во внутриутробном развитии – генетическая предрасположенность. Если хотя бы один из родителей является носителем мутирующего гена, то высока вероятность, что ребенок родится нездоровым. Нахождение матери в зоне повышенной радиации провоцирует возникновение орфанных патологий. Инфекционные заболевания у беременной можно оценивать, как провокатор развития мутаций у ребенка.
Орфанные заболевания: приказ Минздрава
В январе 2014 года Минздравом России был обновлен список орфанных патологий, в который вошло 214 нозологий. Приказом установлено, что ответственность за финансирование медицинской помощи больным возложено на регионы. Перечень обновляется при возникновении на территории государства случаев нового заболевания. Правительством разрабатываются стандарты оказания помощи пациентам с редкими патологиями.


Перечень орфанных заболеваний
Список орфанных заболеваний, встречающихся на территории России, включает следующие категории:
- Микозы: зигомикоз, мукормикоз и т.д.
- Новообразования: тимома, злокачественная саркома мягких тканей и т.д.
- Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм: талассемии, атипичный гемолитико-уремический синдром и т.д.
- Болезни эндокринной системы, расстройства питания и нарушения обмена веществ: гиперпролактинемия, кистозный фиброз и т.д.
- Психические расстройства и расстройства поведения: синдром Ретта и т.д.
- Болезни нервной системы: первичная гиперсомния, гипертрофическая невропатия у детей и т.д.
- Болезни глаза и его придаточного аппарата: наследственные ретинальные дистрофии, атрофия зрительного нерва и т.д.
- Болезни системы кровообращения: первичная легочная гипертензия, желудочковая тахикардия и т.д.
- Болезни кожи и подкожной клетчатки: саркома мягких тканей, болезнь Дюринга и т.д.
- Болезни органов дыхания: гранулематоз Вегенера и т.д.
- Болезни органов пищеварения: язвенный колит желудка и т.д.
- Болезни костно-мышечной системы и соединительной ткани: синдром Маджид, синдром дуги аорты и т.д.
- Болезни мышц: фибродисплазия оссифицирующая прогрессирующая и т.д.
- Болезни мочеполовой системы: наследственные формы азооспермии, нефротический синдром и т.д.
- Врожденные аномалии переднего сегмента глаза: аниридия и т.д.
- Врожденные аномалии, деформации и хромосомные нарушения: болезнь Гиршпрунга, поликистоз почек прогрессивный и т.д.
Какие заболевания встречаются чаще
На территории Российской Федерации чаще встречаются «сиротские» болезни, которые возникают из-за генетических мутаций. Правительством утвержден перечень из 24 заболеваний, из них лечение семи самых дорогостоящих финансируется из бюджета России. Статистика утверждает, что самыми распространенными патологиями считаются: муковисцидоз, гемолитико-уремический синдром, хронический слизистый кандидоз, болезнь Гоше, гипофизарный нанизм.
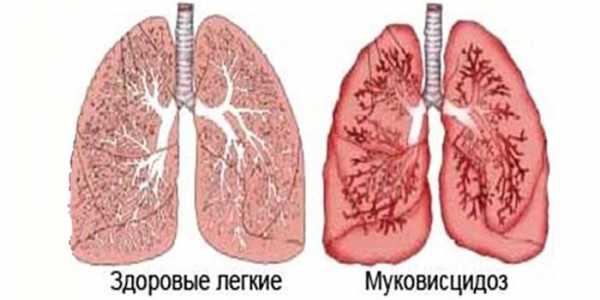
Муковисцидоз
Наследственная болезнь, которая в тяжелой форме поражает железы наружной секреции – муковисцидоз. Патология возникает из-за мутации гена, который регулирует транспорт ионов натрия и хлора через сеточную мембрану клеток. При муковисцидозе поражаются все органы, которые выделяют слизь. Происходит накапливание густого, вязкого содержимого, вывод которого затруднен. Нарушается вентиляция и кровоснабжение легких, возможно появление злокачественных опухолей. У пациентов наблюдается задержка в росте, увеличение печени, вздутие живота, сухой кашель.

Гемолитико-уремический синдром
ГУС – это редкая патология, развивается у детей до трех лет. К причинам возникновения гемолитико-уремического синдрома относят осложнение ДВС-синдрома после перенесенных острых инфекционных заболеваний, системных соединительнотканных болезней. Болезнь провоцирует прием различных медикаментов, осложнения во время беременности, наследственность.
Процедуру диагностики проводят с помощью общего анализа мочи, гистоморфологического исследования. ГУС протекает в три стадии: продромальная, период разгара, период реконвалесценции, либо конец жизни пациента. Каждая стадия имеет особенные проявления, но также выделяют симптомы, которые проявляются на протяжении всей болезни:
- гемолитическая анемия;
- тромбоцитопения;
- острая почечная недостаточность.
Хронический слизистый кандидоз
Генетическое редкое заболевание кожи, слизистых половых органов, оболочек ротовой полости – хронический кожно-слизистый кандидоз. Возбудителями являются дрожжеподобные грибки рода Candida Albicans. Болезнь могут определять на основании микроскопического исследования кожи в мокром препарате с гидроксидом калия. Возникает у детей до 18 лет. Грибки могут подавлять иммунитет, проявляются зудящие высыпания на кожи. Симптоматика хронического слизистого кандидоза очень разнообразна, проявляется бессистемно.
Лечение
Большинство редких заболеваний неизлечимы, поэтому главные цели терапии – повышение иммунитета пациента, улучшение состояния здоровья. Методы лечения разрабатываются на государственном уровне, идет постоянная разработка новых методик, препаратов. Для больных, которым необходимы органы для пересадки, создаются донорские базы. В каждой стране есть список патологий, лечение которых финансируется государством.
Орфанные лекарственные препараты
Орфанные препараты разрабатываются для лечения «сиротских» болезней. Назначение такого статуса является политическим вопросом, государства обеспечивают поддержку и стимулируют разработку такого рода медицинских препаратов. Для стимуляции активности процесса производства орфанных препаратов, некоторые статистические требования могут снижаться. Государство стимулирует разработчиков особыми льготами, снижением налогов, субсидированием разработок, увеличением сроков эксклюзивности на рынке.


Когда следует обратиться к врачу
Многие из нас оттягивают посещения врача, боясь обнаружить страшный диагноз. Следует помнить, что лучше начинать процесс лечения на ранней стадии болезни, это улучшает результат терапевтических процедур, может значительно улучшить качество жизни больного. Если вы заметили симптомы какого-либо отклонения в работе организма, сразу обращайтесь к медицинскому работнику. Лучше доктор опровергнет диагноз, чем болезнь дойдет до непоправимой стадии.
Видео: редкие заболевания
 В ожидании чуда: жизнь с орфанными заболеваниями Смотреть видео
В ожидании чуда: жизнь с орфанными заболеваниями Смотреть видео
Внимание! Информация, представленная в статье, носит ознакомительный характер. Материалы статьи не призывают к самостоятельному лечению. Только квалифицированный врач может поставить диагноз и дать рекомендации по лечению, исходя из индивидуальных особенностей конкретного пациента.
Нашли в тексте ошибку? Выделите её, нажмите Ctrl + Enter и мы всё исправим! Рассказать друзьям:Редкие заболевания — Википедия
Редкие заболевания, орфанные заболевания (англ. rare disease, orphan disease) — заболевания, затрагивающие небольшую часть популяции. Для стимуляции их исследований и создания лекарств для них (орфанные препараты) обычно требуется поддержка со стороны государства.
Многие редкие заболевания являются генетическими, и, следовательно, сопровождают человека в течение всей жизни, даже если симптомы проявляются не сразу. Многие редкие болезни возникают в детстве, и около 30 % детей с редкими заболеваниями не доживают до 5 лет.[1]
Не существует какого-то единого уровня распространенности заболевания в популяции, при котором его начинают считать редким. Заболевание может быть редким в одной части мира или среди какой-то группы людей, но при этом часто встречающимся в других регионах или среди других групп людей.
Не существует единого, широко принимаемого определения редких заболеваний. Некоторые определения полагаются на количество людей, живущих с заболеванием, другие могут включать иные факторы, например, доступность лечения болезни или возможность облегчения её течения.
В США Акт о редких заболеваниях (Rare Disease Act) 2002 года определяет редкие болезни как «болезни или состояния, затрагивающие менее 200000 людей в США» («any disease or condition that affects less than 200,000 persons in the United States»), или примерно 1 человека из 1500. Такое определение очень близко к определению из Закона об орфанных препаратах («Orphan Drug Act») от 1983, федерального закона, написанного, чтобы стимулировать исследования редких болезней и разработку лекарств от них.
В Японии редкие болезни определяются как болезни, затрагивающие менее 50000 пациентов в Японии, или около 1 среди 2500. Однако European Commission on Public Health определяет редкие болезни как угрожающие жизни или хронические серьёзные болезни, имеющие настолько низкий уровень в популяции, что для их изучения и борьбы с ними требуются специальные общие усилия «life-threatening or chronically debilitating diseases which are of such low prevalence that special combined efforts are needed to address them.» Низкий уровень в популяции обычно соответствует менее чем 1 из 2000. Болезни, не являющиеся угрожающими жизни, серьёзными хроническими, либо адекватно излечимыми, исключаются из определения.
В России редкими предлагается считать заболевания с "распространенностью не более 10 случаев на 100 000 человек".[2][3]
В медицинской литературе приняты сходные определения с уровнем распространенности от 1 из 1000 до 1 из 200000.
В список орфанных болезней в России Минздравсоцразвития первоначально внес 86 заболеваний. Количество россиян с этими болезнями оценивалось в чуть менее чем 13 тыс. человек[4]
Однако постепенно перечень таких заболеваний расширялся и по состоянию на 7 мая 2014 года Минздравом РФ в список включены 215 заболеваний[5].
Организация EURORDIS оценивает, что существует от 5 тысяч до примерно 7 тысяч различных редких заболеваний. Хотя для каждого из них встречаемость в популяции будет низкой, совокупно редкими болезнями болеет от 6 до 8 процентов жителей Евросоюза.
Среди различных популяций встречаемость редких болезней будет различна, т.о болезнь, редкая в одной популяции может быть часто встречаемой среди других популяций. Особенно это характерно для генетических и инфекционных заболеваний. Например, муковисцидоз (кистозный фиброз) — генетическое заболевание, редкое для многих частей Азии, но достаточно частое в Европе и бывших европейских колониях. В малых странах или популяциях Эффект основателя может привести к тому, что болезнь, редкая в большинстве мировых популяций, становится очень частой для данного сообщества. Многие инфекционные заболевания часто встречаются на какой-либо определенной территории и редки в других местах планеты. Иные виды заболеваний, например, редкие формы рака, не имеют каких-то неоднородностей в встречаемости и являются просто редкими. Например, любые формы рака у детей считаются редкими, поскольку рак развивается у небольшого количества детей.
Большая часть редких болезней — генетические, и, следовательно, хронические. EURORDIS оценивает, что как минимум 80% редких болезней имеет связанные с ними генетические отклонения.
С 2008 года последний день февраля объявлен днём редких заболеваний.[6]
Орфанные препараты — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 12 февраля 2018; проверки требуют 8 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 12 февраля 2018; проверки требуют 8 правок.Орфанные препараты (англ. orphan drug) — фармацевтические средства, разработанные для лечения редких заболеваний, которые условно называются орфанными («сиротскими») болезнями. Назначение орфанного статуса заболеванию и любым лекарственным препаратам, разработанным для его лечения, является политическим вопросом во многих странах. Поддержка исследований редких болезней со стороны правительств привела к прорывам в медицине, которые не могли бы быть достигнуты в рамках ныне существующей системы финансирования фармакологических исследований. В США и Европейском союзе облегчен вывод орфанных лекарств на рынок и существуют иные финансовые стимулы, например, более длительный период эксклюзивной продажи.
Закон об орфанных препаратах (англ. Orphan Drug Act, ODA), принятый в январе 1983 в США, под влиянием Национального комитета по редким заболеваниям (англ. National Organization for Rare Disorders) и других организаций[1], призван поощрять фармацевтические компании, разрабатывающие лекарственные средства для редких заболеваний (имеющих небольшой рынок). Согласно закону, компании, занимающиеся разработкой препаратов для лечения заболевания, которым страдают менее 200000 человек в Соединенных Штатах, могут продавать их без конкуренции в течение семи лет и могут получать стимулирующие налоговые льготы.[2]
В Европейском союзе (ЕС) был принят аналогичный закон («Regulation(EC) No 141/2000»), в котором фармацевтические средства, разработанные для лечения редких заболеваний, упоминаются как «сиротские лекарственные средства» (англ. orphan medicinal products). Определение, которое даёт ЕС орфанным препаратам, является более широким, чем в США, поскольку в нем также рассматриваются некоторые тропические болезни, которые в первую очередь встречаются в развивающихся странах.[3] Статус орфанного средства предоставляет 10 лет эксклюзивных прав на территории ЕС.[4] Законодательством ЕС по редким заболеваниям занимается Комитет по орфанным препаратам (Committee on Orphan Medicinal Products) Европейского Медицинского Агентства (EMEA).
Законы об орфанных препаратах существуют также в Австралии, Сингапуре и Японии.[5]
В рамках действия законов ЕС и США было разработано множество орфанных препаратов, в том числе препараты для лечения глиомы, множественной миеломы, кистозного фиброза, фенилкетонурии, интоксикации змеиными ядами, идиопатической тромбоцитопенической пурпуры, хронического миелолейкоза и многих других. В США с января 1983 года по июнь 2004 года в общей сложности 1129 различных наименований орфанных препаратов были предоставлены в Управление по разработке орфанной продукции (OOPD) и 249 орфанных препаратов получили разрешение на продажу. Для сравнения, за 10 лет до 1983 года на рынок поступило менее десяти таких продуктов.
С 1983 года по май 2010 FDA утвердила 353 орфанных препарата и предоставила орфанный статус 2116 компонентам. По состоянию на 2010 год уже созданы препараты для лечения 200 болезней из официального списка в 7 тысяч редких заболеваний.[6]
Тем не менее некоторые критики сомневаются в том, что законы об орфанных препаратах были реальной причиной этого увеличения (утверждается, что многие из новых препаратов служили для лечения болезней, которые уже так или иначе были исследованы, и для них разрабатывались препараты вне зависимости от изменений в законодательстве) и на самом ли деле ODA стимулирует производство неприбыльных лекарств. Закон также получил некоторую критику за поддержку ряда фармацевтических компаний, имеющих прибыль от препаратов, которые занимают небольшой рынок, но по-прежнему продаются по высокой цене.[2]
Орфанные препараты, как правило, проходят те же этапы тестирования, что и любая другая фармацевтическая продукция: изучается их фармакокинетика и фармакодинамика, дозирование, стабильность, безопасность и эффективность. Тем не менее некоторые статистические требования снижены в попытке сохранить темпы разработки. Для орфанных препаратов правила допускают, например, что III фаза клинических исследований часто проводится менее чем на 1000 пациентов, поскольку вследствие малой распространённости заболевания требуемое количество просто невозможно набрать.
Поскольку рынок для любого препарата с такими ограниченными возможностями применения по определению не может быть большим и в значительной степени убыточен, часто необходимо государственное вмешательство, чтобы мотивировать производителя обратиться к разработке орфанных препаратов.
Вмешательство правительства с целью поддержки в разработку орфанных препаратов может осуществляться в различных формах[7]:
- Налоговые стимулы и льготы;
- Упрощённая процедура допуска;
- Государственное субсидирование разработки;
- Увеличение срока эксклюзивности на рынке.
Орфанные заболевания. Что это такое, перечень, классификация, лечение
Затрагивающие небольшую часть населения патологии, такие как орфанные, относятся к заболеваниям, для лечения которых требуется поддержка со стороны правительства. Эти болезни связаны непосредственно с генетикой человека. Многие из них проявляются в детстве и сопровождают его в течение всей жизни.
Содержание записи:
Что такое орфанные заболевания?
Орфанные заболевания – это такие патологии, единого определения для которых в настоящий момент еще нет. Часть из них опирается на общее количество людей, которые на постоянной основе борются с основными симптомами. Другая часть определений учитывает доступность компенсационных возможностей для облегчения состояния больного человека.
В 1983 г. был принят Закон об орфанных препаратах. Его создание было предусмотрено с целью стимулирования исследовательских интересов широкого круга ученых. В конечном итоге он может стать причиной, из-за которой будут найдены или созданы способы полного восстановления больных людей.
В разных странах частота регистрируемых случаев для отдельных патологий довольно сильно отличается:
- Япония – 1 случай из 2500;
- США – 1 человек из 1500;
- Россия – 1 среди 10000.
В литературе принята общая статистика, которая считает отдельные типы заболеваний орфанными. По ее данным показатель находится в диапазоне от 1 из 1000 до 1 из 200000 человек.
Типы орфанных заболеваний
По закону в России такими патологиями изначально считались 86 патологий. Постепенно в их число были включены и другие. Количество орфанных заболеваний возросло до 215 в мае 2014 г.
В их число входят такие отклонения, с которыми абсолютное большинство жителей никогда не сталкивались в своей жизни, например, можно привести следующие болезни:
- саркомы костей, хрящевых и мягких тканей;
- раковые новообразования в глазах;
- онкология колоректальной зоны;
- различные типы лимфом;
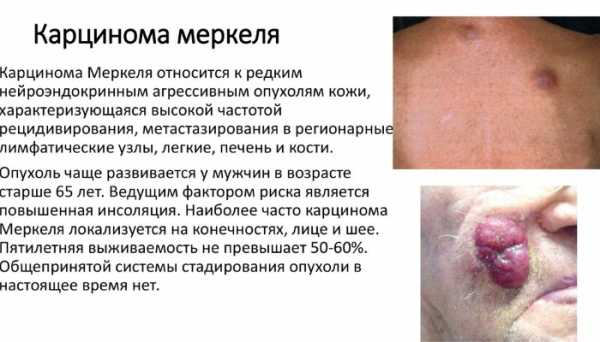
- карцинома Меркеля;
- миеломы;
- лейкозы.
Поскольку перечисленные заболевания в целом имеют похожую природу, но из-за имеющихся названий каждого конкретного типа все они вошли в число орфанных – редко регистрируемых патологий.
Причины
Орфанные заболевания – это ряд патологий, основная часть которых проявляется только в период взросления. В разных странах статистика значительно отличается. Во многом это связано с уровнем жизни местных жителей.
Например, в Индии количество больных людей значительно превышает аналогичный показатель в Европейских странах. Также на развитие болезней оказывает влияние генетика человека. Первые подозрительные симптомы могут быть обнаружены еще в младшем детском возрасте.
К причинам относятся следующие негативные факторы:
- высокий уровень радиационного фона;
- плохая экологическая обстановка;
- загрязнения окружающей среды;
- ослабленная иммунная система;
- наследственные факторы.
С учетом перечисленных причин бороться с риском их развития и улучшать статистику необходимо на высших уровнях со стороны правительства и основных действующих структур.
Причины у детей
Основной фактор – генетическая предрасположенность в сочетании с перенесенными вирусными инфекциями у матери в периоде вынашивания ребенка. Под влиянием этих причин нездоровый ген постепенно мутирует, после чего появляются первые симптомы – повод для серьезного обследования ребенка.
Перечень орфанных заболеваний
Орфанные заболевания – это опасные патологии, с которыми чаще всего врачи сталкиваются в детском возрасте пациента. Поэтому при появлении любых подозрений у часто болеющего ребенка можно предположить носительство генов, ответственных за развитие следующих болезней:
- муковисцидоз – самое распространенное системное отклонение, вызываемое мутацией специфического гена, трансмембранного регулятора муковисцидоза, результатом чего становится почти полная закупорка альвеол и дыхательная недостаточность, частота – 100:105;
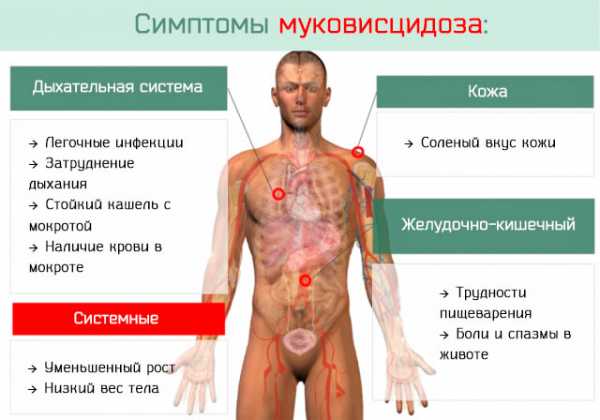
- гемолитико-уремический синдром – при развитии заболевания появляются симптомы гемолитической анемии, тромбоцитопении и азотемии, к ним относятся диарея с кровью, боли в животе, бледная кожа, снижение количества мочи, отклонения в ЦНС и внутренних органах, частота – 1:105;
- пароксизмальная ночная гемоглобинурия – на фоне некоторых причин у больного начинают распадаться эритроциты в составе крови, при этом уровень гемоглобина увеличивается до такой степени, что организм запускает выведение избытка путем почечной фильтрации, частота – 0,55:105;
- апластическая анемия – при ее развитии угнетается функция кроветворения в красном костном мозге, в итоге наблюдается значительное снижение всех типов клеток в составе периферической крови, частота – 0,4:105;
- синдром Стюарта-Прауэра – наследственный недостаток фактора Х в плазме крови, белок под названием гамма-глобулин в норме содержится в ней на уровне 10 %, при падении ниже 1 % результатом становится летальный исход по причине утраты способности крови сворачиваться, частота – 0,2:105;
- тромбоцитопения пурпура – патология, сочетающая активное разрушение тромбоцитов до уровня менее 150х109 шт./л крови, в результате часто наблюдаются длительные и трудно останавливаемые кровотечения, частота – 24,6:105;
- дефект в системе комплемента – патология затрагивает комплекс белковых ферментов, которые требуются для нарушения мембран вредных микроорганизмов, поэтому симптомы схожи с недостатком иммуноглобулинов типа G, частота – 0,5:105;
- преждевременное половое созревание – при наличии такого состояния появление вторичных половых признаков, изменения скелета и другие проявления у девочек наблюдаются еще до 8 лет, у мальчиков – до 7 лет, частота – 4,0:105;
- фенилкетонурия – нарушения обмена ароматических аминокислот, фенилаланина и тирозина, клиническими симптомами которых становятся неврологические и психические расстройства вплоть до умственной отсталости и развитие микроцефалии в будущем, частота – 4,0:105;

- лейциноз – болезнь «кленового сиропа» относится к врожденным нарушениям обменных процессов, при которых в организме накапливаются 3 аминокислоты, лейцин, изолейцин и валин, в результате задерживается развитие, затем наступает фаза угнетения ЦНС вплоть до летаргии, частота – 15,6:105;
- нарушения обмена жирных кислот – при таком состоянии наблюдается дефицит синтеза карнитина, необходимого для переноса жирных кислот в митохондрии для дальнейшего окисления, симптоматически проявляется выраженной гипогликемией из-за резкого увеличения потребления глюкозы всеми тканями организма, частота – 2-10:105;
- гомоцистинурия – нарушения в системе обмена аминокислоты метионина приводят к поражениям нервной системы, зрительного аппарата, костей и суставов, множественному сосудистому тромбозу, частота – 0,4:105;
- тирозинемия – при этом заболевании накапливается недостаточность активности фумарилацетоацетат-гидролазы, в результате чего нарушается обмен тирозина, повреждаются ткани печени и почек, периферическая нервная система, отдаленно патология заканчивается циррозом печени и последующим летальным исходом, частота – 0,05:105;
- глютарикацидурия – в тканях организма происходит накопление глутаровой и 3-OH-глутаровой кислот, которые оказывают отравляющее действие на ЦНС, в частности, на структуры головного мозга под его корой, частота – 0,4:105;
- галактоземия – патология, при которой нарушаются обменные процессы в цепочке превращения галактозы в глюкозу из-за мутационных преобразований структурного гена, в итоге галактоза начинает разрушать ткани печени, нервной системы и хрусталик глаза, частота – 6,6:105;
- болезнь Фабри-Андерсона – отклонения в синтезе фермента альфа-галактозидазы А, который необходим для стабильной работы практически всех клеток, приводят к неизбежным, множественным повреждениям организма, частота – 1,75:105;
- болезнь Нимана-Пика – синдром, при наличии которого происходит накопление жира в различных органах, в частности, страдает печень, селезенка, головной мозг и лимфатические узлы с высоким риском летального исхода, частота – 0,85:105;
- различные ацидемии – метаболические расстройства, при которых наблюдается чрезмерное закисление крови, нарушение нормального обмена аминокислот, в результате которого происходит их накопление в организме, частота – 2-10:105;
- мукополисахаридоз – обменные сбои в процессе синтеза и утилизации гликозаминогиканов провоцируют развитие и накопление дефектов в костях, хрящах и соединительных тканях, частота – 0,6-1,3:105;
- печеночная порфирия – патология, при которой нарушается обмен пигментов в сочетании с увеличением концентрации порфиринов в крови и мягких тканях, может проявляться фотодерматитами, проблемами с кроветворением, расстройствами психики и функций ЖКТ, частота – 10,1:105;
- болезнь Вильсона – неправильный обмен меди приводит к тяжелейшим поражениям периферической и центральной нервной системы и патологическим изменениям в структуре внутренних органов, циррозу печени, частота – 5,84:105;
- незавершенный остеогенез – при таком отклонении не происходит полноценного формирования костных тканей, что в последствии становится причиной повышенной хрупкости всех костей в организме, частота – 6,5:105;
- юношеский артрит с системным началом – ювенильный идиопатический артрит начинается с лихорадочного состояния в течение минимум 3-х суток, после чего к симптомам добавляются эритема, сыпь, генерализованное увеличение лимфатических узлов, плевриты и перитониты, частота – 4,2:105;
- первичная легочная гипертензия – заболевание сопровождается повышением кровяного давления внутри легочной артерии и усилением общего сопротивления в сосудах, среди симптомов наблюдается нехватка воздуха, тахикардия, нарушения сознания, боли за грудиной и другие проявления, частота – 0,4:105.
Орфанные заболевания, встречающиеся с приведенной частотой, являются усредненными данными. Год от года они меняются под влиянием генетических комбинаций при рождении детей и в результате естественной убыли населения. Это всегда влияет на статистику, которая собиралась ранее.
Орфанные заболевания и инвалидность
Подобные заболевания в ряде случаев становятся причиной для признания ребенка инвалидом. Однако в некоторых регионах и при наличии разной симптоматики у 2 детей или взрослых людей с одинаковым диагнозом результат будет полностью зависеть от решения медицинской комиссии.
Поэтому при наличии желания получить инвалидность потребуется выполнить несколько условий:
- собрать все необходимые справки и заключения;
- пройти всех рекомендованных специалистов;
- освидетельствовать результат в МСЭК.
По окончанию всех операций на руки будет выдана справка об инвалидности с указанием сроков ее действительности.
Некоторые люди и, особенно, родители маленьких детей волнуются о том, что статус инвалида помешает дальнейшей социализации. По факту подтвержденная группа нетрудоспособности гарантирует прохождение специальной реабилитационной программы и другие преимущества перед здоровыми людьми:
- подключение к кабельному телевидению на льготных условиях;
- дополнительные скидки на различные виды товаров и услуг;
- лояльные условия для поступления в учебные заведения;
- получение необходимых лекарственных средств;
- снижение тарифов на городскую телефонию;
- санатарно-курортный отдых и лечение;
- бесплатный городской транспорт;
- льготы на предоставление жилья;
- бесплатный доступ в интернет.
В случае ухода за ребенком-инвалидом родителям также предоставляются некоторые преимущества:
- пособие по уходу в размере 60 % от утвержденной минимальной оплаты труда;
- зачисление всего времени, проведенного с ребенком, в трудовой стаж.
Указаны основные критерии, которые остаются практически неизменными. Ряд других преимуществ периодически пересматривается правительством и местными властями, поэтому уточнять все детали рекомендуется непосредственно в региональных ведомствах.
Лечение
Орфанные заболевания – это такие патологии, для лечения которых предназначаются так называемые лекарства-сироты. Эти препараты используются для компенсации состояний, угрожающих непосредственно жизни больного человека или тогда, когда риск связан с приобретением инвалидности. В ряде стран число подобных больных не должно превышать установленного уровня.
 Препараты для лечения орфанных заболеваний
Препараты для лечения орфанных заболеванийСвязано это с тем, что лекарства-сироты относятся к медицинским препаратам, которые экономически невыгодны ни производителю, ни государству. Но они в полной мере отвечают потребностям населения. По этой причине назначать лекарственное средство и выписывать все сопроводительные документы может только специализированный врач.
В России для лечения тяжелых патологий, несмотря на постоянный экономический кризис, выделяются крупные суммы финансовых средств для закупки медицинских препаратов. Поэтому при недостаточном доходе рекомендуется плотно заняться процедурой и предоставить в Медико-Социальную Экспертную Комиссию полный комплект подтверждающих документов.
Когда следует обратиться к врачу
Самое важное условие для максимально раннего выявления имеющихся нарушений, которые могут проявиться клиническими симптомами спустя несколько лет, — это пройти процедуру полного скрининга в период новорожденности. После этого рекомендуется регулярно посещать весь перечень врачей, которые будут рекомендоваться наблюдающим педиатром.
Это в разы увеличит шансы выявить патологию на доклинической стадии, а значит, и назначенное лечение станет в разы более эффективным. Также это позволяет избежать инвалидизации пациента и сохранить здоровье на уровне, достаточном для ведения полноценной повседневной жизни, включая годы обучения и дальнейшую трудовую деятельность по освоенной специальности.
Такой скрининг по действующим нормативам в России проводят в следующие сроки после появления ребенка на свет:
- у недоношенного – на 7 сутки;
- у доношенного – на 4 сутки.
Более ранний забор образцов крови может привести к получению ложноположительных результатов.
Скрининг позволяет выявить предрасположенность к развитию нескольких заболеваний:
- адреногенитальный синдром;
- врождённый гипотиреоз;
- фенилкетонурия;
- муковисцидоз;
- галактоземия.
Все они при несвоевременной диагностике и без подтверждения носительства соответствующего гена неизбежно вели к инвалидизации ребенка по мере его взросления. В ряде областей России список значительно расширен и может включать до 16 и более направлений. Например, в Москве с 2018 г скрининг проводится для выявления 11 заболеваний, включая перечисленные патологии.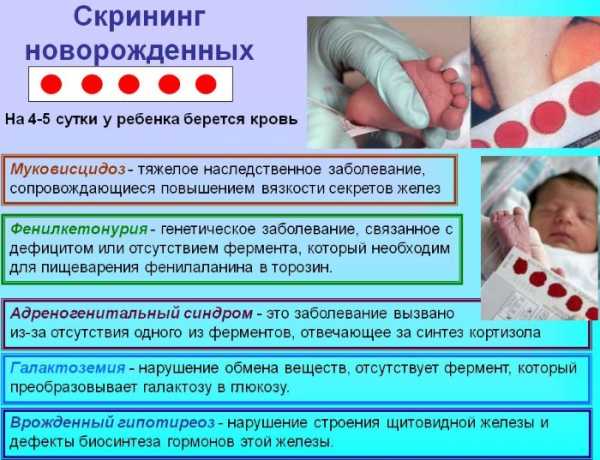
Процедура взятия образца крови не отличается сложностью для специалиста, но может сопровождаться немедленной реакцией ребенка в виде крика в ответ на внезапное ощущение боли.
Взятие крови на анализ именно из пятки обусловлено тем фактом, что пальцы на руках еще слишком малы, чтобы испытывать такие вмешательства. Мать должна спокойно подойти к выполнению необходимой процедуры, поскольку именно от нее во многом будет зависеть, как новорожденный перенесет неприятную манипуляцию.
В некоторых случаях женщины, далекие от медицины, не понимают, зачем делать скрининг, если никто из ближайших родственников не имеет никаких патологий. Необходимость связана с тем, что большинство самых тяжелых генетических заболеваний способны передаваться далеко не через 1 поколение. Поэтому о фактических рисках никто не сможет предоставить реальные данные.
Клинические рекомендации
Основной принцип терапии у больных с подтвержденными орфанными заболеваниями – патогенетическое лечение. Такой способ направлен на коррекционное восстановление функций поврежденных органов и устранение дефектов, вызванных конкретным геном:
- если ген не выполняет функций, за которые несет ответственность, их необходимо возместить;
- если ген производит ненужные эффекты с образованием ядовитых веществ, их избыток следует выводить.
На примере сахарного диабета выполнение такого принципа выглядит, как снижение уровня глюкозы в крови препаратами для внутреннего приема:
- толбутамид;
- карбутомид;
- глиформин;
- глюкофаж;
- адебит.
Другой вариант – использование препаратов с содержанием инсулина.
| Средства для патогенетического лечения | Типы используемых препаратов |
| Вещества для стабильной работы обмена веществ | Глюкоза, витаминно-минеральные смеси, гормональные таблетки, средства для стабилизации функций печени и препараты для нормального кислотно-щелочного и водно-электролитного баланса. |
| Вещества, оказывающие влияние на центральную нервную систему | Блокады на основе новокаина, барбитураты, нейролептики, успокоительные сборы или таблетки, средства для поднятия тонуса. |
| Детоксиканты и препараты для пищеварения | Гепатопротекторы, средства с содержанием ферментов и экстракта поджелудочной железы, ПНЖК и вещества, усиливающие детоксикационную способность печеночной ткани. |
| Вещества, стимулирующие образование и регулярное выведение мочи | Фуросемид, веро-спиронолактон, мочегонные травы, производные бензотиадизина, пиримидина и птеридина.
|

С иммуннозаместительной целью применяют следующие средства:
- неспецифические иммуноглобулины – 10 % смесь бета- и гаммаглобулинов кровяной сыворотки;
- селоколострин – вещество, выделяемое из молозива, полученного от коров в течение 1 суток после отела.
Иммуноглобулины используют для терапии врожденного иммунодефицита и недостаточного веса. Сероколострин содержит до 98 % этих же веществ, при лечении которыми эффективно стимулируется природная устойчивость организма человека к различным разрушающим воздействиям.
В плане генетического наследования, орфанные заболевания относятся к мало контролируемым типам. Это свидетельствует о том, что каждый человек, а в современном мире и каждый младенец, должен быть максимально обследован, начиная с периода новорожденности.
В таком случае при стабильном получении отрицательных результатов вероятность появления симптомов развития генетического заболевания, которое было включено в программу скрининга, будет стремиться к нулю.
Видео про орфанные заболевания
Что такое орфанные заболевания:
Как получить льготное лечение орфанного заболевания: советы юриста
Орфанными (редкими) считаются такие заболевания, распространенность которых охватывает менее 10 случаев на 100 тысяч человек. В России выделяют два списка орфанных заболеваний. Первый включает 19 нозологий (заболеваний), в том числе один из видов ПИД (первичный иммунодефицит). Пациенты с этими 19 заболеваниями имеют дополнительные основания для льготного лекарственного обеспечения.
Второй список из более 200 нозологий (туда входит и большинство видов ПИД) фактически не предполагает дополнительных льгот для пациентов. Однако это совсем не значит, что у пациентов нет других законных оснований для получения бесплатных лекарств. Такими основаниями могут быть инвалидность, вид заболевания, вхождение препарата в перечень и программу ОМС, а иногда даже возраст и многодетность.
Советами, как быстро получить жизненно необходимые лекарственные препараты по месту жительства, поделилась руководитель правовой службы фонда "Подсолнух" Мария Посадкова. Эти наблюдения основаны на многолетнем опыте.
1. Вам установили диагноз – не забудьте взять выписку из федерального центра или медицинского учреждения по месту жительства для его подтверждения.
Помните, что в случае если препарат противопоказан по инструкции (off-label) и (или) необходим по конкретному торговому наименованию, то вам понадобится еще один медицинский документ – протокол врачебной комиссии о назначении конкретного препарата с аргументированным обоснованием его назначения.
2. Если диагноз установлен в федеральном центре, следующий ваш шаг – подача документов в больницу по месту жительства и инициирование проведения врачебной комиссии для подтверждения назначения.
До сих пор законодатель настаивает на том, что выписка федерального центра носит только рекомендательный характер.
3. Если в закупке все равно отказывают (или ее задерживают), а это подвергает вашу жизнь или жизнь вашего близкого опасности, то самое время обратиться в министерство здравоохранения вашего региона и соответствующую страховую компанию (если препарат включен в перечень ЖНВЛП).
4. Если и предыдущий шаг не помог вам достичь заявленной цели, но при этом все необходимые медицинские документы у вас в наличии, то вы можете смело обращаться в контролирующие органы – прокуратуру и Росздравнадзор региона с жалобой на нарушение вашего права на бесплатное лекарственное обеспечение и (или) на доступность и качество медицинской помощи.
Орфанные заболевания - Редкие Люди
Важная информация, которую необходимо знать
Редкие заболевания, орфанные заболевания (англ. rare disease, orphan disease) — заболевания, затрагивающие небольшую часть популяции. Для стимуляции их исследований и создания лекарств для них (орфанные препараты) обычно требуется поддержка со стороны государства.
В список орфанных болезней в России Минздравсоцразвития первоначально внес 86 заболеваний. Количество россиян с этими болезнями оценивалось в чуть менее чем 13 тыс. человек
Однако постепенно перечень таких заболеваний расширялся и по состоянию на 7 мая 2014 года Минздравом РФ в список включены 215 заболеваний
Организация EURORDIS оценивает, что существует от 5 тысяч до примерно 7 тысяч различных редких заболеваний. Хотя для каждого из них встречаемость в популяции будет низкой, совокупно редкими болезнями болеет от 6 до 8 процентов жителей Евросоюза.
Среди различных популяций встречаемость редких болезней будет различна, т.о болезнь, редкая в одной популяции может быть часто встречаемой среди других популяций. Особенно это характерно для генетических и инфекционных заболеваний. Например, муковисцидоз (кистозный фиброз) — генетическое заболевание, редкое для многих частей Азии, но достаточно частое в Европе и бывших европейских колониях. В малых странах или популяциях Эффект основателя может привести к тому, что болезнь, редкая в большинстве мировых популяций, становится очень частой для данного сообщества. Многие инфекционные заболевания часто встречаются на какой-либо определенной территории и редки в других местах планеты. Иные виды заболеваний, например, редкие формы рака, не имеют каких-то неоднородностей в встречаемости и являются просто редкими. Например, любые формы рака у детей считаются редкими, поскольку рак развивается у небольшого количества детей.
Большая часть редких болезней — генетические, и, следовательно, хронические. EURORDIS оценивает, что как минимум 80% редких болезней имеет связанные с ними генетические отклонения.
(данные сведения опубликованы на сайте Википедия)
Редкий диагноз: что такое орфанные заболевания?
Редкий диагноз: что такое орфанные заболевания?
 Редкий диагноз: что такое орфанные заболевания?
Грипп, аллергия, гастрит – к этим заболеваниям мы привыкли к самого детства. Однако существуют и другие, редкие болезни, которые в медицине называются орфанными.
Редкий диагноз: что такое орфанные заболевания?
Грипп, аллергия, гастрит – к этим заболеваниям мы привыкли к самого детства. Однако существуют и другие, редкие болезни, которые в медицине называются орфанными.
Орфанные, или «сиротские», заболевания представляют собой группу редких болезней. На данный момент описано около 7 000 их разновидностей.
Орфанные заболевания встречаются у небольшой части населения, их распространенность составляет около 1 : 2 000 и реже. Данная статистика весьма условна, так как одно и то же заболевание может быть редким в одном регионе и частым в другом. Например, проказа часто встречается в Индии, но редко в Европе.
Откуда берутся орфанные болезни?
Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. В то же время более 50% редких заболеваний проявляются уже во взрослом возрасте.
Реже встречаются токсические, инфекционные или аутоиммунные «сиротские» болезни. Причинами их развития могут быть наследственность, ослабление иммунитета, плохая экология, высокий радиационный фон, вирусные инфекции у мамы и у самих детей в раннем возрасте.
Большинство орфанных заболеваний – хронические. Они в значительной мере ухудшают качество жизни человека и могут стать причиной летального исхода. Для большинства таких болезней не существует эффективного лечения. Основа терапии таких больных – улучшение качества и увеличение продолжительности жизни пациентов.
В настоящее время в развитых странах ведется активное изучение орфанных заболеваний. Оно затрудняется малым количеством пациентов, недостаточным для проведения полноценного исследования. Однако на базе научных изысканий синтезируются новые препараты и выстраиваются схемы лечения больных.
Орфанные заболевания в России
В России редкими предложено считать заболевания с «распространенностью не более 10 случаев на 100 000 человек».
В список орфанных болезней специалисты Минздравсоцразвития РФ в 2012 году внесли 230 наименований, однако в случае выявления новых болезней список будет пополняться. По данным Формулярного комитета Российской академии медицинских наук (РАМН), россиян с этими болезнями насчитывается около 300 тысяч человек.
По данным того же Минздрава, в настоящее время разработаны 24 стандарта оказания помощи больным с редкими заболеваниями, угрожающими жизни и приводящими к инвалидности. Это болезни, которые лечатся, причем терапия направлена на устранение не симптомов, а патологического процесса. Основная часть финансовой нагрузки по лечению орфанных больных из нового списка ляжет на региональные бюджеты.
Орфанные препараты представляют собой лекарственные средства, разработанные
для лечения редких (орфанных, «сиротских») заболеваний. Терапия данными препаратами
требует больших финансовых затрат, поэтому, как правило, финансируется государством.
Неонатальный скрининг
В рамках национального проекта «Здоровье» все новорожденные проходят диагностику (неонатальный скрининг) на 5 наследственных заболеваний: адреногенитальный синдром, галактоземию, муковисцидоз, фенилкетонурию, врожденный гипотериоз. В первые дни жизни по капельке крови, взятой из пятки младенца, проводится исследование на эти наследственные заболевания.
Самое распространенное орфанное заболевание в России
Одним из наиболее распространенных среди редких генетических болезней в нашей стране является муковисцидоз (кистозный фиброз). Он представляет собой системное наследственное заболевание, обусловленное мутацией гена, влияющего на проницаемость клеточных стенок. В результате у больных все секреты (слизь), выделяемые различными органами, имеют повышенную вязкость и густоту, что нарушает работу многих систем организма: дыхательной системы, поджелудочной железы, печени, потовых желез, слюнных желез, желез кишечника, половых желез.
Шанс на полноценное будущее
Муковисцидоз – одно из немногих генетических заболеваний, поддающихся лечебной коррекции. При регулярном применении современных средств: панкреатических ферментов, муколитиков, антибиотиков, гепатопротекторов и витаминов, проведении профилактических мероприятий и кинезитерапии больные муковисцидозом живут полноценной жизнью. Средняя продолжительность ее постоянно растет и достигает сейчас в развитых странах 50 лет (в России она пока вдвое ниже).
Больные муковисцидозом могут создавать семьи и иметь здоровых детей. Но если носителями дефектного гена являются оба родителя, вероятность рождения у них больного ребенка составляет 25%.
Большинство редких заболеваний начинают проявлять себя в детском возрасте.
По статистике, около 30 % детей с редкими заболеваниями не доживают до 5 лет.
Эксперт: Галина Филиппова, врач-терапевт, кандидат медицинских наук
Автор: Олеся Бутузова, врач-педиатр
Список орфанных заболеваний - Редкие Люди
Редкие заболевания, орфанные заболевания — заболевания, затрагивающие небольшую часть населения.
80% редких заболеваний являются генетическими, и, следовательно, сопровождают человека в течение всей жизни, даже если симптомы проявляются не сразу. Многие редкие болезни возникают в детствеи около 30 % детей с редкими заболеваниями не доживают до 5 лет. Так, по экспертным оценкам, летальный исход для 10% больных наступает в возрасте до пяти лет, а еще для 12% — в возрасте от 5 до 15 лет; инвалидность наступает в 65% случаев.
На сегодняшний день понятие орфанных заболеваний закреплено законодательно во многих странах, однако везде в него вкладывается несколько разный смысл.
В США Акт о редких заболеваниях (Rare Disease Act) 2002 года определяет редкие болезни, как «болезни или состояния, затрагивающие менее 200 000 людей в США», или примерно 1 человека из 1500.
В Японии редкие болезни определяются как болезни, затрагивающие менее 50000 пациентов в Японии, или около 1 человека из 2500.
В России редкими предлагается считать заболевания с «распространенностью не более 10 случаев на 100 000 человек». По экспертным оценкам, в России число таких больных составляет около 1,5 млн. человек.
В Европе редкими заболеваниями страдают порядка 40 миллионов человек, в том числе в Германии около 4 миллиона.
Эти болезни характеризуется большим разнообразием патологических состояний и симптомов, что часто приводит к постановке неверного и ложного диагноза.
Значение своевременной диагностики редких заболеваний чрезвычайно велико, поскольку точный диагноз наследственной патологии становится основой адекватного лечения. В России проводится скрининг новорожденных на 6 редких генетических заболеваний, в США проводят скринингноворожденных почти на 50 редких болезней.
Для своевременной диагностики редкого заболевания необходим соответствующий уровень знаний и настороженности врачей (прежде всего первичного звена), а также возможность верификации диагноза с помощью специальных методов исследований.
Учитывая, что до 80% редкой патологии имеет генетически обусловленную природу, генетические исследования должны быть доступны для жителей РФ.
Помощь пациентам с орфанными заболеваниями – обязанность социально ориентированного государства
– Сергей Иванович, расскажите, пожалуйста, о ситуации с редкими заболеваниями в России? Сколько больных зарегистрировано, отражает ли официальная статистика реальное положение дел?

– Распространенность орфанных заболеваний в Российской Федерации ничем не отличается от ситуации в европейских странах или США. По статистике, порядка 5% населения страдают врожденными или наследственными болезнями, которые могут проявиться как с момента рождения, так и в любом возрасте. Большая их часть относится к орфанным заболеваниям. Конечно, они имеют различную клиническую картину. Если мы говорим о тяжелых заболеваниях, то в России это порядка 400–500 тыс. пациентов.
В нашей стране ведутся регистры редких больных – «Семь высокозатратных нозологий» и «24 нозологии». В последний внесено более 16 тыс. пациентов. Причем если раньше в основном в регистре состояли дети, то сейчас практически поровну дети и взрослые. Данные изменения связаны с двумя причинами. Во-первых, мы стали лучше выявлять редкие заболевания, в том числе у взрослых. А во-вторых, пациенты, которые были внесены в регистр еще в детском возрасте, благодаря ранней диагностике смогли вовремя начать патогенетическое лечение и достигли взрослого возраста.
– Как быстро ставится редкий диагноз в России? Какие проблемы с диагностикой существуют и что предпринимается, чтобы их устранить?
– Скорость постановки диагноза зависит от тех подходов, которые мы используем в работе. Один из самых важных способов – скрининг новорожденных. Он позволяет выявлять больных на доклинической стадии, назначить вовремя диету или терапию лекарственными средствами и избежать инвалидизации пациентов в дальнейшем. В нашей стране в скрининг новорожденных включено пять заболеваний (фенилкетонурия, муковисцидоз, галактоземия, андреногенитальный синдром и врожденный гипотиреоз). На наш взгляд, перечень должен быть расширен – это наша обязанность как социально ориентированного государства. Потому что невыявленные редкие заболевания нередко становятся причиной инвалидизации и смерти в младенческом и раннем детском возрасте. Если мы посмотрим на опыт зарубежных коллег, то увидим, что во многих европейских странах скрининг включает до 30 заболеваний, в некоторых штатах США – больше 35.
Если говорить о заболеваниях, которые диагностируются при появлении первых клинических симптомов, то здесь картина не такая радужная. Иногда пациенты идут к своему диагнозу десятилетиями. Это связано с тем, что практикующие врачи часто не думают о том, что речь идет о редком заболевании. Классический пример – болезнь Фабри. От появления первых симптомов до постановки диагноза проходит 7–10 лет. При этом для таких пациентов существует патогенетическое лечение, и если поставить диагноз вовремя, то им можно помочь. Поэтому важна система образования врачей: необходимо доносить информацию, что при нетипичном сочетании симптомов, и неэффективности проводимой терапии, наличии орфанных заболеваний в семейном анамнезе, необходимо подключить генетика к обследованию пациента.
В диагностике по клиническим проявлениям могут помочь также селективные скрининги по группам заболеваний. Такой подход актуален в тех случаях, когда симптоматика может указывать сразу на несколько диагнозов.
Например, для того чтобы подтвердить или опровергнуть болезнь Помпе, которую можно спутать с другими поясно-конечными мышечными дистрофиями, на базе нашего медико-генетического научного центра внедряется диагностическая панель на 82 миопатии. ДНК-диагностика доступна и бесплатна для всех пациентов в России.
– Насколько отличаются условия диагностики в крупных и небольших городах? Как взаимодействуют региональные и федеральные медицинские центры в данном вопросе?
– Условия для клинической диагностики редких заболеваний есть как в федеральных, так и региональных центрах, но для всей страны актуальна проблема низкой настороженности врачей. Другое дело лабораторная диагностика наследственных орфанных болезней с помощью генетических методов. Она доступна в федеральных медико-генетических центрах (в Москве и Томске), в которых можно получить консультацию врача-генетика и генетическую диагностику. Важно, что эти услуги бесплатны для пациента.
На мой взгляд, это оптимальная схема, когда диагностику, а также инициирование лечения осуществляют именно крупные или федеральные медицинские центры: у них больше опыта работы с редкими заболеваниями да и с организационной и экономической точки зрения это гораздо выгоднее, чем открывать специализированные центры диагностики редких болезней по всей стране. На Экспертном совете 9 ноября мы как раз обсуждали систему оказания помощи пациентам с лизосомными болезнями накопления. И в очередной раз пришли к выводу, что она должна иметь несколько уровней – на некоторых из них могут быть задействованы федеральные центры (диагностика и начало терапии, селективные скрининги), а на других – региональные (наблюдение пациентов и лечение).
– Какие возможности терапии предусмотрены в системе здравоохранения РФ для пациентов с редкими заболеваниями? С какими сложностями сталкиваются больные?
– У нас существует федеральная программа «Семь нозологий», включающая такие заболевания как: гемофилия, муковисцидоз, гипофизарный нанизм, болезнь Гоше, злокачественные новообразования лимфоидной, кроветворной и родственных им тканей, рассеянный склероз, состояния после трансплантации органов и тканей, которую уже смело можно называть «12 нозологий» – в нее недавно были дополнительно внесены 5 заболеваний: атипический гемолитико-уремический синдром, ювенильный артрит и мукополисахаридоз I, II, VI типов. Благодаря этому пациенты с такими диагнозами имеют право получать дорогостоящую терапию бесплатно за счет федерального бюджета. Это очень важно, потому что региональные бюджеты в большинстве своем не позволяют обеспечить финансирование лечения в полной мере.
Существует также программа «24 нозологии», в соответствии с которой лечение редких заболеваний, вошедших в этот список, должно финансироваться из региональных бюджетов. К сожалению, не во всех регионах программа работает бесперебойно. Мы встречаемся с ситуациями, когда пациенту удается получить лечение только через суд.
Иногда сталкиваемся с тем, что не все препараты для лечения редких заболеваний, зарегистрированные в мире, приходят на наш рынок своевременно. И несмотря на то, что у нас существует процедура ускоренной регистрации орфанных препаратов, необходимо обеспечивать их скорейшее включение в госпрограммы, а также развивать программы по обеспечению «орфанных» пациентов незарегистрированными в РФ препаратами по жизненным показаниям.
– Насколько система поддержки пациентов с редкими заболеваниями в России отличается от зарубежных? Есть ли какие-то уникальные практики в России?
– Мы регулярно обмениваемся опытом работы с зарубежными коллегами и можем отметить уникальность нашей системы диагностики и лекарственного обеспечения пациентов, работающей на федеральном уровне, принимая во внимание большой масштаб страны. Благодаря слаженной работе Департамента лекарственного обеспечения и регулирования обращения медицинских изделий Минздрава и его руководителя Елены Анатольевны Максимкиной федеральная программа «Семь нозологий» работает «как часы».
– Лечение орфанных заболеваний дорогостоящий процесс. Как распределяются траты при диагностике и терапии между пациентом и государством? Что поможет оптимизировать расходы?
– Диагностика редких заболеваний для российских пациентов бесплатна. Она осуществляется в региональных и федеральных центрах и состоит из двух этапов. Первый – это клиническая диагностика, когда заподозрить наличие редкого диагноза должен врач амбулаторной практики. Затем необходима лабораторная диагностика. Она проводится федеральными и крупными региональными центрами, которые также могут осуществлять первичную консультацию пациентов. Ежегодно такое обследование проходят 18 тыс. человек.
Далее пациенты с подтвержденным диагнозом получают направление в федеральные центры для повторного обследования и подтверждения диагноза. Здесь им назначают лечение и в некоторых случаях инициируют его. Далее оно продолжается в региональных центрах – в рамках программ «Семь нозологий» и «24 нозологии».
Сам пациент вряд ли сможет оплатить свое лечение, поскольку оно очень затратное.
Оптимизировать этот процесс можно, на мой взгляд, передав вопросы финансирования лекарственного обеспечения всех редких пациентов на федеральный уровень. Централизованные закупки через Минздрав России более эффективны – они дают возможность договариваться с производителями ЛС о снижении цены и фиксировать ее на государственном уровне. Что поможет получить препараты для большего количества пациентов по более низкой цене.
– Какие дальнейшие изменения в законодательстве помогут улучшить ситуацию с диагностикой редких заболеваний?
– Важно расширить программы массового скрининга новорожденных и ввести селективный скрининг групп высокого риска. Наша медицина носит профилактическую направленность, мы должны выявлять редкие заболевания еще до проявления клинических симптомов. Но, к сожалению, сейчас из-за низкого уровня осведомленности об орфанных заболеваниях на уровне органов управления мы сталкиваемся с ситуациями, когда в отдельных регионах скрининги не проводят вообще. Очень важно, чтобы у региональных структур Минздрава России и медучреждений было понимание, что отмена скрининга – это нарушение федерального закона.
– Сегодня активно идет работа по лекарственному обеспечению пациентов с редкими заболеваниями. Также специалисты настаивают на расширении списка заболеваний для неонатального скрининга. Какие критерии для этого могут быть использованы, какие заболевания должны быть включены в первую очередь?
– Существуют определенные принципы для включения новых нозологий в программы скрининга, разработанные ВОЗ. Основные: заболевание должно быть часто встречаемым, должны существовать методы для его диагностики, а также препараты для лечения. Раньше считалось, что лечение должно быть экономически оправданно, но сейчас эта парадигма меняется на «если пациенту можно помочь – это необходимо сделать». Например, В США рассматривается вопрос введения неонатального скрининга на спинально-мышечную атрофию. Лечение этого заболевания очень дорогое, но, я уверен, таких пациентов в США будут лечить. А во многих штатах уже внедрен скрининг на лизосомные болезни накопления. Например, на те же болезни Фабри и Гоше.
В нашей стране важно включать в скрининги в первую очередь те заболевания, для лечения которых у нас за последнее время были зарегистрированы препараты – это болезнь Помпе, дефицит Альфа-1 антитрипцина, семейная гиперхолестеринемия, некоторые формы наследственного амилоидоза, ДЛКЛ (дефицит лизосомной кислой липазы).
– В последнее время как в профессиональном сообществе, так и среди широкой аудитории орфанные заболевания обсуждаются все чаще. С чем это связано?
– На мой взгляд, это связано с достижениями нашей медицины и системы здравоохранения в вопросах профилактики, диагностики и лечения наиболее часто встречающихся редких заболеваний. Ведь, как правило, именно такие успехи и те изменения, которые они за собой влекут, становятся причиной обсуждения той или иной темы. Также, конечно, в этом велика роль пациентских организаций.
– Говоря о роли последних – в чем именно она проявляется, когда мы говорим об осведомленности общества о редких заболеваниях?
– Я вижу большую роль пациентских организаций в формировании социогуманитарного подхода в политике государства. Для того, чтобы в обществе было понимание того, насколько серьезен этот вопрос и, что редкое заболевание может коснуться каждой семьи, необходима серьезная образовательно-информационная работа. Именно пациентские организации могут рассказать о том, как орфанное заболевание влияет на жизнь человека, его семьи и в последствии на все общество. Привлечь внимание к тому, что редким больным можно помочь, если вовремя поставить диагноз и начать лечение. Фактически они помогают сформировать запрос со стороны общества для дальнейшего развития службы медицинской помощи. Кроме того, пациентские организации оказывают информационную, а часто моральную, психологическую и социальную поддержку пациентам с редкими заболеваниями и их семьям.
Редкие заболевания | Орфанные заболевания
Редкими (орфанными) называют те заболевания, которые затрагивают небольшое количество людей от общей численности населения планеты. При этом единого показателя распространенности редких заболеваний в популяции не существует. В различных странах и частях света уровень встречаемости отличается. Например, в Европе болезнь Хансена (проказа) выявляется крайне нечасто, а в центральной Африке ее диагностируют как у детей, так и взрослых. Муковисцидоз распространен в европейских странах, тогда как в Азии это генетическое заболевание является довольно редким.
К тому же в трактовку редких заболеваний могут входить и другие факторы: доступность лечения и возможности симптоматической терапии. Поэтому в различных уголках мира уровень заболеваемости редкими патологиями определяют по-разному. Так, в Японии редкими считаются те болезни, которые встречаются у 1 человека из 2,5 тыс. населения, в Америке этот показатель составляет 1 из 1,500 тыс. людей, Европе 1:2000 и в России 5 человек на 50 тыс. населения. Согласно статистике, более 60 млн. людей земного шара страдают редкими заболеваниями, а это около 8 % жителей планеты. Со стороны каждого государства требуется огромная поддержка для научно-медицинских исследований и разработки препаратов для лечения редких болезней.
По инициативе Европейской организации по редким заболеваниям EURORDIS дата 28 февраля или 29-е в високосный год во всем мире получила статус международного дня редких (орфанных) заболеваний. Именно последний день зимы символизирует тот факт, что редкие болезни сегодня стали серьезной медицинской проблемой, требующей особого внимания со стороны людей и государства.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Редкие генетические заболевания
Среди всех редких заболеваний большинство приходится на генетические или врожденные болезни, которые имеют хроническое течение и проявляются у детей в разные периоды жизни. Большинство редких генетических патологий манифестируют в младенчестве и сопровождаются расстройствами зрения и слуха, выраженными изменениями внешности, нарушениями двигательной и психической функции. При этом вследствие прогрессивного течения генетической болезни более 25 % детей не доживают до 4-5 лет.
Наследственные заболевания у детей характеризуются неблагоприятным прогнозом. Однако своевременное лечение редких генетических болезней в большинстве случаев дают хорошие результаты, врачам удается улучшить качество жизни маленьких пациентов и увеличить показатели выживаемости.
Что такое орфанные заболевания
Термины «орфанные заболевания» или «болезни-сироты» (от англ. orphan — сирота) появились в 1983 году, когда в Америке был принят закон о препаратах-сиротах («Orphan Drug Act»), благодаря которому поощрялись фармацевтические компании за разработку лекарственных средств с целью лечения редких патологий. В России на эту проблему обратили внимание только в 2011 году и закрепили права пациентов с редкими заболеваниями в законе «Об охране здоровья граждан».
Сегодня понятие орфанных (редких) болезней закреплено в медицинской терминологии законодательством многих стран. В группу орфанных болезней входят малоизвестные генетические, аутоиммунные и инфекционные патологии, а также редкие формы рака, аномалии и синдромы. Орфанные заболевания, вызванные мутациями генов, характеризуются тяжелым хроническим течением и требуют длительного дорогостоящего лечения. Другие виды редких патологий также ухудшают качество жизни, быстро прогрессируют, могут приводить к инвалидности и смертельному исходу. Большинство орфанных болезней у детей и взрослых являются малоисследованными состояниями, для которых еще не разработаны методы терапии. Кроме того, не часто удается диагностировать подобные заболевания своевременно.
Откуда берутся орфанные заболевания
Генетические орфанные болезни вызваны мутациями ДНК, то есть хромосомными нарушениями. Такие заболевания могут передаваться из поколения в поколение, к примеру, по аутосомно-рецессивному типу, когда оба родителя несут дефектный ген, или по аутосомно-доминантному типу наследования, при котором у одного из родителей есть генетический дефект. И в том и другом случае вероятность передачи мутантного гена ребенку составляет 50 %.
Редкие аутоиммунные заболевания возникают в результате того, что иммунная система начинает атаковать собственные ткани и органы. Однозначной причины этому процессу нет. Орфанные болезни детей и взрослых также могут быть спровоцированы малоизученными вирусами и бактериями, а также токсическим воздействием. Происхождение большинства орфанных заболеваний у взрослых и детей на сегодняшний день остается не известным. Так, несмотря на многочисленные исследования, этиология чрезвычайно редкого синдрома Роххад до сих пор не выяснена.
Сколько редких заболеваний существует
По статистике, в мире насчитывается более 8000 тысяч видов орфанных заболеваний, из которых 2/3 в основном поражают детей. Для 6 тыс. редких заболеваний не разработаны протоколы лечения. Еженедельно в медицинской литературе описывают несколько новых болезней. Но официальное определение патологий основывается на этиологических данных, клиническом описании и принятой классификации. Перечень орфанных заболеваний разделяют на отдельные нозологические формы, а также на группы болезней, имеющие общие названия и признаки.
Какие заболевания встречаются чаще
Чаще всего встречаются генетические орфанные болезни — 80-85 % случаев. Показатели заболеваемости варьируются в зависимости от географического фактора. Среди наиболее распространенных орфанных патологий следует отметить: гемофилию, муковисцидоз, болезнь Гентингтона, синдром Туретта, болезнь Альцгеймера, мышечную дистрофию, синдром Хантера, а также различные формы рака (нейробластому, карциному Меркеля, миелоидный лейкоз и т.д.).
Орфанные заболевания у детей
Более половины орфанных болезней диагностируют у детей. В основном у маленьких пациентов выявляют врожденные или генетические заболевания, симптомы которых могут не проявляться длительное время. Также дети разных возрастов нередко страдают редкими онкологическими и инфекционными болезнями. Своевременная диагностика генетических детских заболеваний является благоприятным фактором прогноза.
Лечение редких заболеваний (методы)
Большая часть редких заболеваний — это неизлечимые тяжелые патологии, угрожающие жизни. Но сегодня существует ряд эффективных методов, позволяющих поддерживать нормальное качество жизни, а также достигать выживаемости в течение многолетнего периода. На сегодняшний день разработано около 450 лекарственных средств, устраняющих одно из звеньев патогенеза. Орфанные препараты применяют для лечения редких заболеваний, которые могут привести к инвалидности или угрожают жизни пациента.
Более 95 % редких заболеваний не лечатся. Однако генетики сегодня проводят доклинические исследования по новым технологиям лечения онкологических и генетических патологий. Это методы РНК-интерференции и редактирование генома.
Диагностика и лечение редких заболеваний в Москве
Из-за отсутствия лабораторно-технической базы и достаточного числа квалифицированных специалистов во многих регионах, российские пациенты нередко сталкиваются с проблемой своевременной и точной диагностики редких заболеваний. Однако сегодня в стране функционируют несколько федеральных медцентров, в том числе и в Москве, где есть все условия для проведения тщательной диагностики при подозрениях на орфанные болезни.
Сейчас каждый ребенок после рождения проходит неонатальный скрининг на более 5 редких заболеваний, включая генетические патологии. Верификация диагноза выполняется ведущими генетиками в современных лабораториях.
Вопрос лечения редких болезней является нерешенным на уровне отечественной медицины, поскольку большинству орфанных пациентов необходимые оригинальные препараты не доступны. В России занесено в Федеральный реестр около 17. тыс. человек, которые находятся под постоянным наблюдением специалистов. Если для редкой болезни не существует эффективного лечения, врачи поддерживают состояние больных и увеличивают продолжительность их жизни при помощи специализированного питания и орфанных препаратов.
Диагностика и лечение редких заболеваний в Израиле
Для выявления редких заболеваний специалисты Израиля используют инновационные генетические тесты, безошибочно обнаруживая известные мутации на ранней стадии развития. Ведущие эксперты в данной области медицины не только устанавливают точный диагноз, но и лечат орфанные болезни, применяя новейшие методы энзимозаместительной и генной терапии. Исключительную эффективность при многих генетических патологиях доказала трансплантация костного мозга, проводимая всемирно известными гематологами и хирургами Израиля.
Помощь фондов при лечении редких генетических заболеваний
Не каждое государство располагает достаточными бюджетными средствами для поддержки пациентов с орфанными заболеваниями. В таких случаях помощь оказывают благотворительные фонды, которые сотрудничают с лучшими клиниками мира и финансируют детей или взрослых с редкими болезнями на всех этапах диагностики и лечения. Например, благотворительные фонды «Алеша», World Vita и Русфонд оказывают неоценимую финансовую поддержку детям, страдающим орфанными заболеваниями. Если у вас возникли вопросы, оставьте контактную заявку на сайте или позвоните по телефону горячей линии.
Нередкая ситуация с редкими болезнями / Здоровье / Независимая газета
Согласие на финансирование при полном сопротивлении сторон
 Больше половины больных орфанными заболеваниями – дети. Фото c сайта www.mos.ru
Больше половины больных орфанными заболеваниями – дети. Фото c сайта www.mos.ru
Орфанные заболевания называют также сиротскими. Сироты и есть – так или иначе они оказываются последними в очереди на государственную заботу. Только два года назад очередь дошла и до них – понятие «пациенты с орфанными заболеваниями» появились в правовом поле с принятием Закона «Об охране здоровья граждан России».
К орфанным отнесли болезни, которые имеют распространенность не более 10 случаев на 100 тыс. человек. Согласно официальной статистике, 300 тыс. жителей России страдают орфанными заболеваниями, общий перечень которых состоит из 230 позиций. Однако неофициально называются другие цифры – от 1,5 до 5 млн человек. Такой разброс данных объясняют новизной понятия, несовершенством диагностики, скрининга и отсутствием учета пациентов, страдающих редкими болезнями.
Дискуссии по поводу орфанных заболеваний идут у нас давно, но четкого понимания и определенного подхода к больным так и не выработано. Постановлением № 403 от 26 апреля 2012 года был утвержден «Перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности». В перечень редких заболеваний, поддающихся терапии, вошли 24 нозологии. Но федеральный регистр лиц, страдающих этими заболеваниями, до сих пор полноценно не ведется. С этими заболеваниями сложно все, начиная с точной постановки диагноза, что по плечу не каждому специалисту. В 80% случаев в основе болезни лежит генетическая патология.
Болезни, входящие в группу редких, – тяжелые, страдающие ими пациенты без лечения долго или хотя бы полноценно не проживут. А лечение дорогое. По закону финансирование орфанных заболеваний было возложено на региональные бюджеты. Эксперты считают, что, даже если бы субсидирование было полноценным, децентрализация орфанной программы сразу сделала ее неэффективной. А субсидирование, как легко догадаться, далеко не полное. По данным Минздрава, необходимыми лекарствами обеспечены всего 63% больных. По данным пациентских организаций, в некоторых субъектах РФ необходимое лечение получает не более 20% нуждающихся в нем.
До сих пор достоверно не известно, какой объем средств требуется на препараты для лечения орфанных заболеваний. Не все лекарства даже зарегистрированы на нашей территории. Иногда врачи на свой страх и риск ввозят остро необходимые жизнеспасающие средства из-за границы.
Прошло два года после того, как орфанные заболевания получили официальный статус. Некоторые итоги были подведены в докладах на информационном семинаре для профессиональных СМИ.
Президент Национальной ассоциации организаций больных редкими заболеваниями «Генетика» Светлана Каримова представила мониторинг качества жизни, медицинской и социальной помощи пациентам на основании круглых столов, проведенных в 18 регионах страны. Она рассказала о неравенстве доступа лечения для пациентов. В ряде регионов не только отказываются обеспечивать их лекарствами, но даже не включают в федеральный регистр больных с орфанными заболеваниями. Наиболее затратные заболевания – пароксизмальная ночная гемоглобинурия (заболевание крови), атипичный гемолитико-уремический синдром (характеризуется механической гемолитической анемией, тромбоцитопенией и поражением почек) и мукополисахаридоз различных типов (заболевание соединительной ткани, которое приводит к различным дефектам костной, хрящевой, соединительной тканей). Ни один региональный бюджет не в состоянии обеспечить их терапию. Практически пациенты остаются один на один с болезнью. Приходится добиваться необходимых лекарств через суд.
Елена Красильникова, руководитель Центра изучения и анализа проблем народонаселения, демографии и здравоохранения Института ЕвроАзЭс, представила результаты масштабного исследования оказания медицинской помощи и лекарственного обеспечения больных редкими заболеваниями. Исследование проводилось в 2012–2014 годах в 63 регионах Российской Федерации. Выявлены и описаны основные проблемы, а также мифы и заблуждения, связанные с редкими болезнями.
Так, пациенты с орфанными заболеваниями – вовсе не обязательно люди с ограниченными возможностями. Они становятся инвалидами только при несвоевременно начатом или нерегулярном лечении. Не все редкие заболевания дорогостоящие. Орфанные болезни – это главным образом организационная проблема. Необходимо не только лекарственное обеспечение, но высокотехнологичные диагностические центры, лечебные учреждения, где такие пациенты лечатся амбулаторно или стационарно, и в первую очередь медицинские регистры и базы данных.
Больше половины больных редкими болезнями – дети. Отчего случился генетический сбой, в результате которого возникла болезнь, неизвестно. Может произойти с каждым. Депутаты Госдумы, видимо, считают, что их детей это не коснется. Иначе трудно объяснить, почему они отклонили законопроект, в соответствии с которым средства на лечение орфанных заболеваний должны были выделяться из федерального бюджета. А члены Совета Федерации даже заявляли ранее о намерении в пять раз увеличить финансирование лечения редких болезней.
Мало того, президент Владимир Путин в мае подписал предложение по софинансированию расходов бюджетов субъектов РФ на орфанные заболевания за счет ассигнований из федерального бюджета. И срок определил – 14 июля. Родители больных детей с надеждой ждали этого. Но, видимо, какая-то Баба-яга против. И за месяц до определенного президентом срока пришло решение Госдумы – оставить все как есть. Какие там дети, не до них, есть расходы поважнее.
Комментарии для элемента не найдены.