Самые редкие болезни в мире фото
Редкие болезни становятся частыми - МК
Кому нужно обследоваться на редкие
Многие ученые называют наступивший век веком наследственных заболеваний. Количество детей с генетическими патологиями стремительно растет. Впрочем, редкие болезни существовали в мире всегда, но говорить о них начали лишь в конце прошлого века. Потому что появились новые диагностические возможности, ученые научились секвенировать геном и находить в нем специфические поломки, отвечающие за то или иное нарушение. Так в наш лексикон вошли слова «муковисцидоз», «мукополисахароз» и многие другие.
За последние 15 лет ученые узнали причины более 7000 заболеваний, более 2,5 тысячи расстройств и синдромов. Раньше пациенты с редкой болезнью просто погибали без возможности получить диагноз и необходимую медицинскую помощь. Сегодня неонатальный (у новорожденных) и пренатальный (в утробе матери) скрининги, предымплантационная диагностика при ЭКО предоставляют широкие возможности: например прервать беременность и совершить другую попытку; воспользоваться донорскими клетками или сперматозоидами; выбрать из всех эмбрионов ЭКО здоровый для подсадки в матку; начать раннее лечение. «Например, если в семье уже есть ребенок с мукополисахаридозом, вероятность того, что другие дети тоже будут болеть, составляет от 5о% для мальчиков (пр болезни Хантера, мукополисахаридозе второго типа), до 25% - для мальчиков и девочек при всех других типах — говорит профессор, главный научный сотрудник НИКИ «Научно-исследовательский клинический институт педиатрии им. Ю.Е.Вельтищева» Алла Семячкина. — Тогда при наступлении беременности проводится дородовая диагностики на 11-й неделе (берется биопсия плода). Если плод поражен, родители могут решить, прерывать беременность или нет».
Сегодня в нашей стране можно диагностировать абсолютно любое редкое заболевание из известных. И все же, как считает заведующая лабораторией наследственных болезней обмена веществ ФГБНУ Медико-генетического центра, председатель Совета экспертов Всероссийского общества редких (орфанных) заболеваний Екатерина Захарова, тотальное генетическое обследование всех пар и всех новорожденных на все генетические заболевания бессмысленно: «Наследственных болезней очень много, однако делать скрининг логично на то, что можно предупредить и лечить. Потому что может получиться так, что знание станет печалью».
Доктор Захарова приводит пример, как недавно молодая пара, планирующая ребенка, решила провести полное генетическое тестирование. У женщины обнаружили две мутации в гене, ответственном за позднюю форму дегенеративного заболевания ЦНС, которое развивается после 35 лет и приводит к необратимой инвалидизации. «Хотели ли они это знать? Может, им нужно было просто родить ребенка, который был бы стопроцентно здоровым? Но женщина впала в депрессию и перестала думать о продолжении рода. Поэтому российский скрининг новорожденных проводится только на те заболевания, от которых есть лечение», — продолжает Захарова.
Кроме того, проходить диагностику для выявления носительства генов, вызывающих развитие тяжелых наследственных заболеваний, врачи рекомендуют людям, в роду которых были кровнородственные браки.
В нашей стране новорожденным проводят ранний скрининг на галактоземию, фенилкетонургию, муковисцидоз, врожденный гипотериоз и андрогенитальный синдром Дауна. Список, безусловно, будет расширяться. Некоторые регионы уже сделали это. Так, в Приморском крае в качестве эксперимента детей начали обследовать сразу на 40 патологий, для 29 из которых сегодня существует эффективное лечение, в Москве скрининг новорожденных проводятся на 11 заболеваний.
Кроме того, диагностические методы постоянно совершенствуются. Так, сегодня появился генетический скрининг, определяющий синдром Дауна и другие хромосомные аномалии у плода со 100%-ной вероятностью по анализу крови матери — уже с 4-й недели беременности в ней появляются фрагменты плодного ДНК.
С редким можно быть счастливым
Диагностика редких болезней особенная. Эксперты говорят, что иногда, хотя и нечасто, диагноз можно поставить с первого взгляда по особым симптомам, присущим только этой болезни. Но чаще бывает иначе. Диагностическая одиссея может длиться годами и вылиться в определение болезни лишь спустя 5–10 лет после начала ее симптомов.
Генетики любят рассказывать историю, как была открыта болезнь фенилкетонурия. Это случилось в Англии. Молодая пара обратилась к врачу по поводу двоих детей, которые родились совсем обычными, а через несколько лет у них начались нарушения интеллектуального развития. Кроме того, от них очень плохо пахло, мышами, — у отца на этот запах даже началась аллергия.
Врачи долго не понимали, что с ними. Было понятно лишь то, что болезнь наследственная. Один из докторов начал проводить простые тесты с мочой пациентов. И однажды сделал тест с хлоридом железа — жидкость вдруг позеленела. Несколько месяцев лаборатория пыталась выделить вещество, которое придавало такую окраску, пришлось исследовать 40 ведер биоматериала. И вещество нашли. Теперь с помощью особого теста исследуют всех новорожденных в мире, а лечить болезнь элементарно: особой диетой, которая предотвращает повреждения интеллекта и позволяет вести обычный образ жизни. Сегодня пациентов с фенилкетонурией в России обеспечивают специальным питанием за госсчет.
Снежана Митина, глава пациентской организации «Хантер-синдром», тоже долго не знала, что с ее ребенком. Он родился настоящим красавцем и богатырем, больше 4 килограммов. И до трех лет развивался, как другие дети. А потом начались звоночки.
— Когда сыну удаляли грыжу, у него развилась реакция на наркоз — и нам предложили обследоваться. Тогда и поставили страшный диагноз. Конечно, я не поверила, думала, ошиблись, — ведь дети с синдромом Хантера (мукополисахаридозом 2-го типа) низкорослые, с ярко выраженной внешностью, а мой был высокий красивый блондин. И вообще я считала, что больные дети рождаются у алкоголиков и асоциальных людей. Но постепенные изменения внешности при синдроме начинаются после двух лет, с трех дети перестают расти и вскоре становятся «на одно лицо», превращаясь в заколдованных гномиков. Болеют в основном мальчики, в мире всего две девочки с синдромом Хантера. В 2000 году, когда сыну поставили диагноз, лекарств еще не было. Но в 2006 году появился препарат. В 2008 году мы поехали в Германию за счет Минздрава, где получили лечение, а потом добились, чтобы лекарство было зарегистрировано и у нас. Государство в 2012 году включило мукополисахаридозы в перечень 24 редких болезней, а в 2018-м году в программу «Семь нозологий», по которой такие пациенты обеспечиваются лекарствами.
Такая терапия в нашей стране применяется уже десять лет, и за это время темпы инвалидизации таких пациентов снизились, а продолжительность их жизни выросла. Снежана говорит, что лечение творит чудеса. Они начали принимать лекарства, когда ее ребенок уже перемещался на четвереньках, а по утрам забывал часть слов или каких-то действий. К тому же, говорит Снежана, хантеренки — очень ласковые дети, которые дают гораздо больше любви, чем обычные. Однако ее пациентская организация занимается вопросами лекобеспечения детишек со всеми типами мукополисахаридоза (МПС): «Мне принципиально, чтобы лечение получали все дети».
«Нужно в целом решить проблему с обеспечением пациентов незарегистрированными в стране лекарствами, иначе они не могут получать лекарства за государственный счет», — говорит председатель правления Центра экспертной помощи «Дом редких» Анастасия Татарникова. В некоторых регионах эта проблема решается: например Москва выделяет средства на закупку незарегистрированных препаратов из фонда мэра, в Нижнем Новгороде есть социальные выплаты. Однако необходим единый подход.
«Что такое жить с редким заболеванием? — говорит эксперт «Дома редких» Кирилл Куляев. — В 2010 году, когда мне поставили орфанный диагноз, в мире не было от него ни одного лекарства. Мне давали прогноз — пять лет. Сейчас пошел уже десятый год — и умирать я не собираюсь. Сегодня я постоянно получаю лекарства, живу полноценной жизнью, если не считать походов к докторам. Женился. У меня две дочки».
ххх
Понятие редкости в разных странах законодательно установлено свое. Например, в Европе орфанным считается заболевание, которое встречается 1:2500. В России 1:10 000. В Британии — 1 к 40 000. 80% орфанных пациентов заболевают в возрасте до 15 лет. Большинство редких болезней — наследственные.
В 2011 году главу об орфанных препаратах включили в закон об охране здоровья граждан, тем самым обеспечив таким пациентам право на лечение. «В 2012 году в стране начал формироваться федеральный регистр редких пациентов, — рассказывает руководитель Центра изучения и анализа проблем народонаселения, демографии и здравоохранения Института ЕАЭС Елена Красильникова. — К настоящему моменту число пациентов в регистре удвоилось, а расходы на лекарственное обеспечение редких больных выросли в три с половиной раза».
И все же многие годы за лекобеспечение большинства редких пациентов отвечали регионы, что нередко ложилось непосильным бременем на их бюджеты. Существенно изменит ситуацию перевод таких пациентов под федеральное крыло, что случится совсем скоро. Весной Дмитрий Медведев дал поручение перевести лекарственное обеспечение всех орфанных заболеваний на федеральный уровень.
Во многих случаях ранняя диагностика и начало лечения помогут избежать развития тяжелых осложнений, предотвратить развитие ранней инвалидизации и летального исхода. Не говоря уже об улучшении качества жизни детей и членов их семей. Некоторые редкие болезни лечатся очень просто. Диетотерапией или, например, высокими дозами витаминов. «А вообще с развитием персонализированной медицины скоро каждое заболевание будет считаться редким. Ученые открыли молекулярные механизмы патогенеза рака, и теперь, например, рак легких или рак молочной железы — это десятки и даже сотни разных болезней, каждая из которых требует индивидуальное лечение», — говорит Екатерина Захарова.
И современное здравоохранение уже готово принять этот вызов.
20 самых странных и редких болезней, известных человечеству
В настоящее время во всем мире проживает огромное количество людей, которые страдают одними из самых редких и самых странных болезней, известных человеку. О некоторых из них, вероятно, многие из вас не слышали, но они действительно существуют, и они влияют на повседневную жизнь тех, кому не повезло, что у них появились симптомы. В некоторых случаях есть методы лечения, но не всем так везет. Фактически, у большинства нет известного лечения, и они все еще сбивают с толку врачей.
Синдром взрывающейся головы

Синдром взрывающейся головы звучит как что-то из плохого фильма ужасов, но на самом деле это серьезная болезнь, поражающая тысячи людей во всем мире. Симптомами болезни являются звуки взрыва бомбы, выстрела пистолета, удара тарелок или другой вид громкого шума в голове при попытке уснуть. Однако нет никаких симптомов боли, отека или любого другого физического аспекта. Чаще всего их получают люди старше 50 лет, но и люди в возрасте 10 лет тоже сообщают о таких случаях.
Синдром Алисы в стране чудес

Синдром Алисы в стране чудес звучит как нечто полностью выдуманное, но это не так, хотя он назван в честь романа Льюиса Кэрролла. Симптомы включают галлюцинации, дезориентацию, дисметропсию или искажение размера. Симптомы распространены в детстве, но большинство из них вырастают в подростковом возрасте. Симптомы не вредны и со временем исчезнут.
.
7 редких болезней, которые теперь можно вылечить
В мире очень немногие редкие заболевания поддаются эффективному лечению. Но благодаря этим семеркам это число могло просто снизиться, по крайней мере, немного.
СМОТРИ ТАКЖЕ: РЕДКОЕ ЗАБОЛЕВАНИЕ ВЫЗЫВАЕТ БУКВАЛЬНО ИСКЛЮЧЕНИЕ КОСТИ ЖЕНЩИН
Для того, чтобы болезнь считалась редкой, в США она обычно поражает только около 200000 человек. На сегодняшний день насчитывается около 6800 редких заболеваний, признанных Национальными институтами здравоохранения (NIH).
Но следует иметь в виду, что, хотя каждое заболевание редко, само по себе, совокупное число пораженных людей является значительным. Согласно этой статье, они влияют примерно на 30 миллионов человек , или 1 из 10 в США
Чтобы болезнь считалась редкой в ЕС, она должна поражать менее 5 в каждом 10000 человек.
Некоторые редкие болезни действительно очень редки. Некоторые из них имеют менее дюжины известных случаев, тогда как другие более распространены, такие как рассеянный склероз, муковисцидоз и мышечная дистрофия Дюшенна.
В совокупности считается, что эти расстройства затрагивают 6–7% населения в развитых странах.
Многие медицинские работники предупреждают, что существует острая необходимость в эффективных методах лечения некоторых или всех из них. Но разработка медикаментозного лечения - это не быстрый процесс.
По крайней мере, эти 7 расстройств наконец получили одобренные FDA методы лечения их многострадальных пациентов.
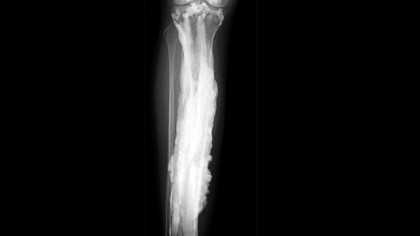
1. Мелореостоз был загадкой в течение многих лет.
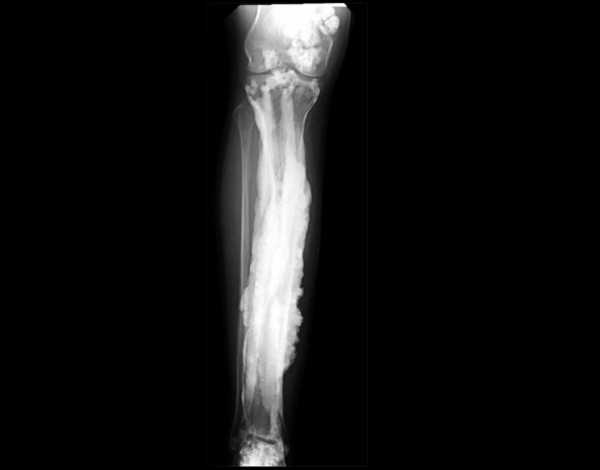 Источник: Национальные институты здравоохранения
Источник: Национальные институты здравоохранения В апреле прошлого года мы сообщили о возможной причине этого редкого заболевания под названием мелореостоз.Заболевание невероятно редкое, на сегодняшний день во всем мире зарегистрировано всего 400 случаев.
После согласованных усилий по поиску причины, Национальный институт здравоохранения, кажется, наконец нашел причину. До этого никто не знал причины.
Тимоти Бхаттачарья, доктор медицины, объясняет: «Ученые ранее предполагали, что генетические мутации, ответственные за мелореостоз, происходят во всех клетках человека с этим заболеванием».
Согласно недавнему исследованию, причиной, по-видимому, является дефектный ген у пациентов.Гены MAP2K1 отвечают за продукцию белков MEK1.
Когда эти гены не функционируют должным образом, создается избыток белка. Это вызывает скопление лишней кости на пораженных участках, что создает проблему.
Это понимание станет важной информацией для создания потенциального лекарства в будущем.
2. Неходжкинская лимфома наконец получила одобренное FDA лечение
Два типа лечения неходжкинской лимфомы, рецидивирующий или рефрактерный грибовидный микоз (MF) или синдром Сезари (SS), недавно получили одобрение FDA .Оба эти заболевания представляют собой злокачественные опухоли Т-лимфоцитов крови.
До сих пор их было невероятно трудно вылечить, но ожидание лекарства теперь может закончиться. Новые препараты, Могамулизумаб-kpkc, вводятся внутривенно и являются первым лекарством, одобренным FDA для лечения заболеваний.
Препарат был одобрен после клинических испытаний с участием более 372 пациентов , которые получали препарат или химиотерапию. Результаты были впечатляющими.
Среди тех, кто получал препарат, их выживаемость после употребления наркотиков была вдвое выше, чем в группе химиотерапии.Хотя это не так долго, почти 8 месяцев по сравнению с 3, это обнадеживает для будущего развития.
Утверждение «удовлетворяет неудовлетворенные медицинские потребности этих пациентов», - сказал Ричард Паздур, доктор медицинских наук, директор Онкологического центра FDA и исполняющий обязанности директора Управления гематологии и онкологических продуктов в Центре оценки и исследований лекарственных средств FDA. в заявлении.
3. Теперь можно лечить наследственный ангионевротический отек (НАО).
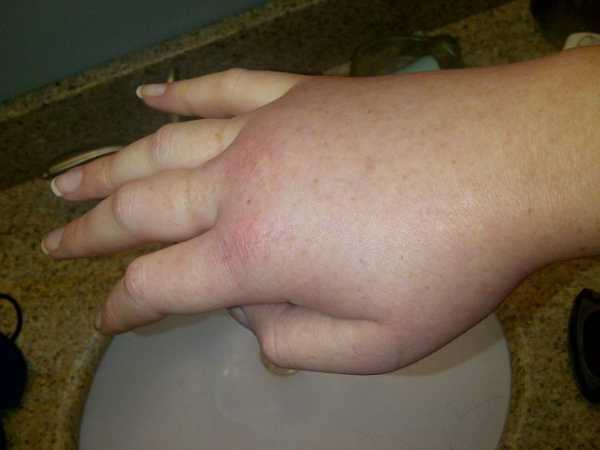 Источник: Пример приступа НАО.Источник: LucyHAE / Wikimedia Commons
Источник: Пример приступа НАО.Источник: LucyHAE / Wikimedia Commons Новый препарат, ланаделумаб-флио, недавно был одобрен FDA для предотвращения приступов наследственной ангиодемы (HAE). Его можно использовать для помощи пациентам старше 12 лет.
HAE - очень редкое генетическое и потенциально опасное для жизни заболевание. Это может вызвать повторяющиеся приступы отека (отека) по всему телу пациента.
HAE в настоящее время поражает примерно от 1 из 10 000 до 1 из 50 000 человек. У детей также есть шанс 50% проявить расстройство, если кто-то из их родителей страдает.
Больные часто будут испытывать невыносимую боль, плохое самочувствие и рвоту из-за набухания стенки кишечника. Если в горле возникает опухоль, это может привести к смерти от удушья.
Новый препарат нацелен на выработку фермента калликреина плазмы, который хронически не контролируется у больных НАО. Он вводится непосредственно под кожу путем самостоятельной инъекции и имеет период полураспада около 2 недель.
4. Болезнь Фабри теперь лечится лучше
Болезнь Фабри - еще одно редкое заболевание и генетическое заболевание, которое вызывает накопление жира в кровеносных сосудах, почках, сердце и нервах пациентов.Этот жир, глоботриаозилцерамид (GL-3), также может накапливаться во многих других органах тела и в конечном итоге является потенциально смертельным.
Проблема вызвана дефицитом фермента, который приводит к скоплению жира по всему телу. Современные методы лечения просто заменяют недостающий фермент, вместо того, чтобы обеспечивать «лечение» как таковое.
Новое лекарство, Мигаластат, было недавно одобрено FDA, и это первое пероральное лекарство, предназначенное для лечения заболевания у взрослых.Он отличается от существующих методов лечения увеличением активности фермента, дефицитного в организме, альфа-галактозидазы A.
Его эффективность была продемонстрирована в ходе 6-месячного плацебо-контролируемого исследования с участием 45 взрослых пациентов. Пациенты, получавшие новый препарат, показали гораздо большее снижение глоботриаозилцерамида в различных органах тела.
Он также был безопасен в ходе 4 клинических испытаний 139 пациентов Fabry .
5.Мы надеемся, что бета-талассемия теперь имеет потенциальное лекарство.
Бета-талассемия - редкое заболевание, которое снижает количество гемоглобина, вырабатываемого в красных кровяных тельцах. Для всех, кто помнит школьные уроки биологии, это белок, который позволяет эритроцитам переносить кислород по телу.
По меньшей мере, жизненно важно сохранить вам жизнь. Больные страдают хронической нехваткой железосодержащего белка в кровотоке, что приводит к кислородному голоданию в некоторых частях тела.
Больные, как правило, страдают анемией из-за нехватки жизнеспособных эритроцитов.
Симптомы часто включают бледность кожи, слабость, утомляемость, а также гораздо более серьезные осложнения. Люди с бета-талассемией подвергаются повышенному риску развития аномальных тромбов.
Новый препарат, Луспатерсепт, который в настоящее время находится в разработке, представляет собой гибридный белок, который регулирует выработку эритроцитов на поздней стадии в костном мозге пациентов.
Тем самым повышается уровень гемоглобина и снижается нагрузка на переливание крови для пациентов и учреждений первичной медико-санитарной помощи.
[см. Также]
6. Боковой амиотрофический склероз (БАС) теперь можно лечить
 Источник: Ассоциация БАС
Источник: Ассоциация БАС Боковой амиотрофический склероз (БАС) - редкое прогрессирующее нейродегенеративное заболевание, поражающее нервные клетки в головной и спинной мозг. Название болезни происходит от сочетания греческого «А», что означает «нет», «Мио», что означает мышцы, и «трофический», что означает питание.
То есть буквально «без питания мышц». Любая мышца, которая не получает питания, истощается или атрофируется.
Латеральный относится к тому факту, что он влияет на спинной мозг человека, где расположены части нервных клеток, которые сигнализируют о мышцах и контролируют их. По мере прогрессирования заболевания пораженные участки страдают от рубцевания и затвердевания, что с медицинской точки зрения называется «склерозом».
Это приводит к отмиранию двигательных нейронов головного мозга и к потере у пациентов двигательной функции, что может привести к потере речи, неспособности есть, потере движения и даже контроля дыхания.
Различные новые испытания могут привести к лечению БАС в недалеком будущем. Они включают генную терапию и подходы с использованием стволовых клеток для лечения БАС.
7. Ювенильный идиопатический артрит теперь может иметь эффективное лечение
Ювенильный идиопатический артрит, ранее называвшийся ювенильным ревматоидным артритом, - редкое заболевание, поражающее детей в возрасте до 16 лет.
Это заболевание приводит к постоянному боль в суставах, опухоль и общая скованность у больных.Некоторые пациенты могут страдать от заболевания всего несколько месяцев, в то время как другие страдают всю оставшуюся жизнь.
В некоторых случаях заболевание может привести к проблемам роста, повреждению суставов и воспалению глаз. Текущее лечение включает в себя контроль боли и воспаления, улучшение функции и предотвращение повреждения суставов.
В настоящее время лекарства нет, и лечение включает использование кортикостероидов, противовоспалительных препаратов, противоревматических препаратов, модифицирующих заболевание (DMARD), и биологических агентов.Хотя большинство из них просто помогают с симптомами, другие, например биологические агенты, могут уменьшить системное воспаление и повреждение суставов в долгосрочной перспективе.
Примеры включают этанерцепт и адалимумаб. Другие методы лечения могут включать биологические агенты, подавляющие иммунную систему пациента.
К ним относятся абатацепт, ритуксимаб, анакинра и тоцилизумаб.
.Это самые редкие (и самые страшные) тропические болезни, связанные с путешествиями
У вас есть шишка под кожей после зарубежной поездки? Это самые редкие (и самые страшные) тропические болезни, связанные с путешествиями
- Доктор Рон Беренс из лондонской больницы тропических болезней раскрывает все
- Он предупреждает о личинках, которые растут под кожей и которые можно увидеть движущимися
- Одна инфекция приводит к «разрушению кожи и мягких тканей»
- ВНИМАНИЕ: Графические изображения
Кэти Эми для MailOnline
Опубликовано: | Обновлено:
Пробежки или похмелье - основные проблемы, связанные с болезнью во время отпуска для большинства путешественников.
Однако, если вы отважитесь отправиться в экзотические страны, вас ждут более зловещие недуги.
Здесь MailOnline Travel с помощью доктора Рона Беренса из Больницы тропических болезней в Лондоне рассматривает некоторые из самых редких болезней, связанных с путешествиями, в том числе личинок, которые растут под кожей, язвы, разрушающие ткани, и инфекцию, которая приводит к до крайнего отека конечностей.
Прокрутите вниз для просмотра видео
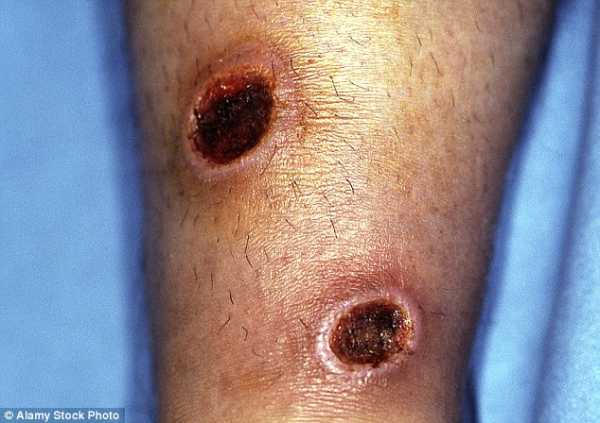
Лейшманиоз - это хроническая кожная язва, передающаяся через песчаную муху, которая затем передается паразиту.
Болезнь: лейшманиоз
Откуда она пришла: во многих частях мира, включая Средиземноморье.Однако в основном он встречается в Южной Америке, Боливии и некоторых частях Амазонки. Также может встречаться в Индии и некоторых частях Африки.
Что это такое: «Это хроническая кожная язва», - сообщает MailOnline Travel доктор Рон Беренс, консультант Национальной службы здравоохранения по тропической и туристической медицине в Больнице тропических болезней в Лондоне. «Он заразился через песчаную муху, которая передается через паразита. Он существует как растущая язва. Есть разные типы, но кожная форма наиболее распространена. Обычно мы наблюдаем в больнице примерно один случай в месяц.

Хотя лейшманиоз может возникать во многих частях мира, он чаще встречается в Южной Америке и Боливии (на фото)
Лечение: «Иногда он исчезает или лечит сам себя, но мы обычно даем больным довольно сильные лекарства, паразит из организма », - объясняет доктор Беренс. «Пациент должен будет приходить ежедневно в течение 21 дня для лечения».
Профилактика: Используйте средство от насекомых с 50% -ным ДЭТА и примите все меры предосторожности, чтобы не быть укушенным.
Болезнь: лихорадка Ласса
Откуда она пришла: Западная Африка
Что это такое: По данным Всемирной организации здравоохранения, это «острое вирусное геморрагическое заболевание», вызываемое вирусом Ласса. Он передается человеку через контакт с продуктами питания или предметами домашнего обихода, загрязненными экскрементами грызунов. Симптомы начинаются с лихорадки и общей слабости, хотя могут последовать головная боль, боль в горле, боль в груди, тошнота, диарея и боль в животе. ВОЗ также указывает, что в тяжелых случаях может развиться «отек лица, жидкость в полости легких и кровотечение изо рта, носа, влагалища или желудочно-кишечного тракта».В случае летального исхода смерть обычно наступает в течение 14 дней от начала заболевания.

Лихорадка Ласса - острое вирусное геморрагическое заболевание, возникшее в Западной Африке (на фото), хотя его часто трудно отличить от таких болезней, как Эбола
.О редких заболеваниях | www.eurordis.org
| Заболевание или расстройство определяется как редкое в Европе, если оно поражает менее 1 человека в 2000 году. Одно редкое заболевание может поражать лишь несколько пациентов в ЕС, а другое затрагивает целых 245 000 человек. Насчитывается более 6000 редких болезней. В целом от редких заболеваний могут пострадать 30 миллионов граждан Евросоюза. 80% редких заболеваний имеют генетическое происхождение и часто являются хроническими и опасными для жизни.
|
Благодарности
Этот видеоклип был подготовлен Animo Productions при бесплатной поддержке Burson-Marsteller Brussels. Представленные пациенты являются членами Ирландского национального альянса пациентов с редкими заболеваниями. Мы благодарим их и компанию Rare Diseases Ireland за их неоценимый вклад. Мы также благодарим Грегуара за разрешение использовать его песню «Toi et Moi» в качестве саундтрека к видео.
.







