Синдром айкарди что это такое
Инфантильные спазмы.Синдром Айкарди - Медицинский справочник
Синдром Айкарди необходимо исключать у девочек с инфантильными спазмами. Кроме инфантильных спазмов у девочек с этим заболеванием выявляется частичная или полная агенезия мозолистого тела, пороки развития грудного отдела позвоночника и хориоретинальные лакунарные дефекты. Поражение глаз часто представлено колобомой зрительного нерва и микроофтальмией. Считается, что синдром сцеплен с Х-хромосомой и проявляется преимущественно у девочек. Описана и другая форма Х-сцепленных инфантильных спазмов, выявляющихся у мальчиков.
Существуют противоречивые мнения о том, может ли вакцина против коклюша играть роль в развитии инфантильных спазмов. Вакцина вводится в том возрасте, на который попадает пик дебюта инфантильных спазмов. Поэтому в большом количестве случаев наблюдается совпадение по времени двух событий — вакцинации и дебюта инфантильных спазмов. Кроме того, точное определение момента дебюта инфантильных спазмов во многих случаях затруднено. Однако в тех случаях, когда зарегистрировано совпадение по времени вакцинации и дебюта инфантильных спазмов, патогенетическая связь между этими событиями вызывает сомнение. Возможно, что причинно-следственная связь все же существует у небольшой части детей, особенно если выраженная реакция со стороны нервной системы возникла в течение 24 часов после иммунизации. Также возможно, что в некоторых случаях вакцина оказывает действие совместно с другими, не идентифицированными факторами, провоцируя дебют клинических проявлений у детей, предрасположенных к развитию заболевания. Применение убитой вакцины позволяет значительно снизить риск побочных эффектов, ассоциированных с вакцинацией.
Тот факт, что инфантильные спазмы возникают у одних младенцев с заболеваниями нервной системы и отсутствуют у других детей с аналогичными заболеваниями, позволяет предположить, что важную роль в развитии инфантильных спазмов играет генетическая предрасположенность.
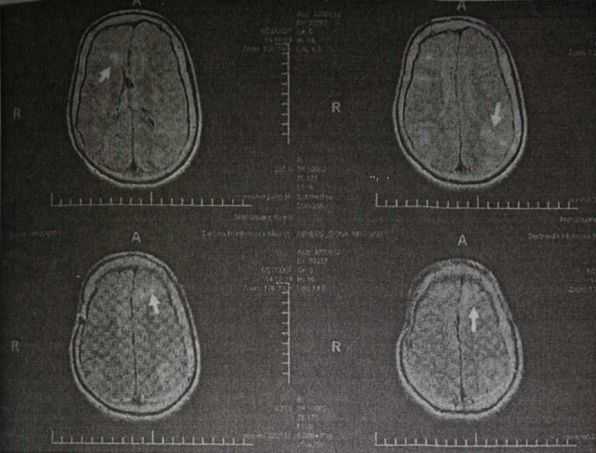
МРТ ребенка с туберозным склерозом, у которого первым симптомом заболевания были инфантильные спазмы. Кортикальные туберсы показаны стрелками
Кроме того, при помощи МРТ возможно диагностировать такие пороки развития мозга, как порэнцефалию, агенезию мозолистого тела или гидранэнцефа- лию. При применении методов нейровизуализации патологические изменения обнаруживаются у 70—80% пациентов с инфантильными спазмами. Наиболее частой аномалией, выявляемой у большого количества пациентов, служит диффузная церебральная атрофия. Так как лечение АКТГ может приводить к развитию транзиторной атрофии вещества головного мозга, рекомендуется проведение МР-исследования до начала терапии АКТГ.
Так как у некоторых детей причиной инфантильных спазмов служит пиридок- синовая зависимость, важным диагностическим тестом, проводимым у пациентов, с не установленным этиологическим фактором, является внутривенное введение пиридоксина в дозе 100—200 мг во время ЭЭГ-мониторинга. При этом у младенцев с пиридоксиновой зависимостью наблюдается улучшение клинической картины (прекращение приступов) и электроэнцефалографических аномалий в течение нескольких минут после введения препарата.
Если причину приступов не удается установить, рекомендуется метаболическое исследование, включающее скрининговое исследование плазмы крови и анализ мочи для выявления аминокислот, исследование плазмы крови для определения уровня аммиака, органических кислот, лактата, пирувата, проведение печеночных тестов. Исследование спинномозговой жидкости должно включать определение уровня глюкозы (необходимо сравнить уровень глюкозы в крови и в спинномозговой жидкости), белка и количества клеток. Исследование спинномозговой жидкости на содержание аминокислот, пирувата и лактата следует проводить при подозрении на метаболическое заболевание. Так как большинство детей получают кортикостероиды, необходимо исследование плазмы крови для оценки уровня электролитов, кальция, фосфора, глюкозы, а также проведение анализа мочи.
Поделитесь ссылкой:Синдром Кернса — Сейра — Википедия
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьезным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии (сокращенно CPEO), синдром, который характеризуется изолированным поражением мышц, контролирующих движения век (поднимающая верхнее веко, круговая мышца глаза) и контролирующих движения глаз (экстраокулярных мышц). Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных: CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия может включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения [1]. В обоих этих заболеваниях, вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, и, конечно является прогрессирующим.
Это триада CPEO, двусторонняя пигментная ретинопатия и блокада сердца была впервые описана в докладе о случае у двух пациентов в 1958 году доктором медицинских наук Томасом П. Кернс, (англ. Thomas P. Kearns) и доктором медицинских наук Джорджем Помероя Сейр (англ. George Pomeroy Sayre).[2] Второй случай был опубликован в 1960 году Ягером и соавторами, которые сообщили об этих симптомах у 13-летнего мальчика.[3]Предыдущие случаи внезапной смерти пациентов с CPEO были опубликованы, как от сердечной аритмии. Другие случаи отмечали особую пигментацию сетчатки, но ни одна из этих публикаций не была документирована как три патологии, возникающие вместе в качестве генетического синдрома.[4] Кернс опубликовал определяющий случай в 1965 году, описывающий 9 несвязанных случаев этой триады.[4] В 1988, была впервые выявлена связь между KSS и крупномасштабными удалениями мышечной митохондриальной ДНК (сокращенно мтДНК)[5][6] После этого открытия, многочисленные делеции в митохондриальной ДНК были связаны с развитием КСС. [7][8][9]
Синдром Кернса — Сейра происходит спонтанно в большинстве случаев. В некоторых случаях была передача по наследству посредством митохондриального, аутосомно-доминантного, или аутосомно-рецессивного наследования. У него нет пристрастия к расе или полу, и нет никаких известных факторов риска. По состоянию на 1992 было всего 226 случаев, зарегистрированных в опубликованной литературе.[10].По состоянию на 2017г. получены данные о том, что при синдроме Кернса – Сейра не прослеживается наследственная передача данной патологии, заболевание регистрируется в виде единичных случаев.
Лица с KSS предстают первоначально со сходными симптомами с типичной CPEO. Начало в первой и второй декадах жизни.
Первым симптомом этого заболевания является односторонний птоз, или проблемы при открытии век, который постепенно прогрессирует и приводит к двустороннему птозу. Когда птоз усиливается, пострадавший обычно запрокидывает шею, поднимая подбородок в попытке предотвратить окклюзию зрительной оси опустившимися веками. Наряду с коварным развитием птоза, движения глаз в конечном итоге становятся ограниченными, в результате чего, лицо больше полагается на поворот головы из стороны в сторону или вверх и вниз для просмотра объектов в периферическом поле зрения.
- Пигментная ретинопатия
KSS приводит к пигментации сетчатки, прежде всего, в задней части глазного дна. Наблюдается диффузная депигментация пигментного эпителия сетчатки с наибольшим эффектом в жёлтом пятне. Этим KSS отличается от пигментного ретинита, где пигментируется периферия. Вид сетчатки при KSS аналогичен тому, которое наблюдалось при миотонической дистрофии типа 1 (сокращенно DM1). Умеренная куриная слепота может наблюдаться у пациентов с KSS. Потеря остроты зрения, как правило, мягкая и происходит только у 40-50 % больных.[11]
- Нарушения сердечной проводимости
Это чаще всего происходит после образования птоза и офтальмоплегии.[11]Атриовентрикулярная блокада (сокращенно «AV») является наиболее распространенным дефицитом сердечной проводимости. Это часто прогрессирует до третьей степени желудочковой блокады, которая является полным блокированием проводимости от предсердия в желудочек. Симптомы сердечной блокады включают обморок, непереносимость физических нагрузок и брадикардию
- Церебральная фолатная недостаточность
У пациентов с синдромом Кернса — Сейра очень часто обнаруживается церебральная фолатная недостаточность - синдром, при котором уровни 5-MTHF в спинномозговой жидкости снижены, несмотря на нормальные уровни фолиевой кислоты и 5-MTHF в плазме крови.[12] Назначение фолиниевой кислоты может в некоторых случаях облегчить симптомы недостаточности и даже скорректировать наблюдаемые на снимках мозга отклонения, особенно если терапия была начата на ранних стадиях заболевания.[13] Предполагаемая причина церебральной фолатной недостаточности у пациентов с синдрмом Кернса-Сейра - дисфункция сосудистого сплетения, нарушающая поступление фолатов в спинномозговую жидкость.[14]
- Другие
Согласно описанию заболевания, представленному Кернс в 1965 году, а также описаниям в более поздних публикациях, некоторые симптомы возникают не у всех пациентов. Среди этих симптомов - слабость мышц лица, глотки, туловища и мышц конечностей, потеря слуха, небольшой рост, электроэнцефалографические изменения, мозжечковая атаксия и повышенный уровень белка в спинномозговой жидкости.
KSS является результатом делеций в митохондриальной ДНК (мтДНК), которые вызывают определённый фенотип. мтДНК передается исключительно от яйцеклетки матери.[15] Митохондриальная ДНК состоит из 37 генов, найденных в одной кольцевой хромосоме размерностью 16569 спаренных оснований в длину. Среди них 13 генов кодируют белки дыхательной цепи переноса электронов(сокращенно «ЭTЦ»), 22 кодируют транспортировку РНК (тРНК) и два кодируют ряд больших и малых субъединиц, которые образуют рибосомные РНК (рРНК). 13 белков, участвующие в ЭТЦ в митохондриях, необходимы для окислительного фосфорилирования. Мутации в этих белках приводит к нарушениям производства энергии в митохондриях. Этот дефицит клеточной энергии быстрее всего проявляется в тканях, которые сильно зависят от аэробного метаболизма, таких как мозг, скелетные и сердечные мышцы, органы чувств и почки. Это лишь один фактор, участвующий в представлении митохондриальных заболеваний.
Есть и другие факторы, влияющие на проявление митохондриальной болезни, кроме величины и расположения мутации. Митохондрии дублируются посредством деления клеток во время беременности и в течение всей жизни. Поскольку мутация митохондриальной болезни чаще всего встречается на ранних сроках беременности при этих заболеваниях, только митохондрии в мутированной линии являются дефектными. Это приводит к неравномерному распределению дисфункциональных митохондрий в каждой клетке, и в различных тканях тела. Это называется гетероплазмия, которая характерна для митохондриальных заболеваний, включая KSS. Распределение мутантных мтДНК в каждой клетке, ткани и органе, зависит от того, когда и где произошла мутация.[16] Это может объяснить, почему два пациента с одинаковыми мутациями в мтДНК могут представлять совершенно разные фенотипы и, в свою очередь различные синдромы, Публикация в 1992 году Fischel-Ghodsian и др. определила удаление одного и того же 4977-б.п. в мтДНК у двух пациентов с двумя совершенно различными заболеваниями. Один из пациентов имел характерный KSS, а другой пациент — совсем другое заболевание, известное как синдром Пирсона.[17] Усложняет дело то обстоятельство, что в некоторых случаях синдром Пирсона, как было выявлено, может прогрессировать внутри KSS позднее в жизни.[18] Более поздние исследования привели к выводу, что дублирование мтДНК может также играть важную роль в определении присутствия фенотипа. Дублирование мтДНК, кажется, характерно для всех случаев KSS и синдрома Пирсона, в то время как они отсутствуют в CPEO.[18][19]
Удаления мтДНК в KSS различаются по размеру (1.3-8kb), а также положению в митохондриальном геноме. Наиболее распространенным удалением является 4.9kb и простирается от позиции 8469 до позиции 13147 в геноме. Это удаление присутствует примерно у ⅓ людей с KSS.
Нейроофтальмологи, как правило, участвуют в диагностике и лечении KSS. Человек должен быть заподозрен в КСС на основе клинических данных экспертизы. Подозрение на миопатию должно быть усилено для пациентов с отсутствием офтальмоплегии и определенного набора параличей черепных нервов (паралич глазодвигательного нерва, паралич блокового нерва, паралич отводящего нерва). Первоначально исследования изображения часто проводится, чтобы исключить более общие патологии. Диагноз может быть подтвержден мышечной биопсией, которая может быть дополнена PCR для определения мутаций мтДНК. Биопсия: Это не обязательно биопсия глазной мышцы, для демонстрации гистопатологические аномалий. Сечение мышечных волокон, трихромное окрашение пятна Гомори рассматривается с помощью световой микроскопии. В мышечных волокнах, с более высоким содержанием мутированных митохондрий, существует более высокая концентрация митохондрий. Это придаёт этим волокнам темно-красный цвет, в результате чего общий вид биопсии описывается как «рваные красные волокна». Аномалии могут быть также продемонстрированы в образцах биопсии мышц с использованием других гистохимических исследований, таких как пятен митохондриальных ферментов, с помощью электронной микроскопии, биохимических анализов мышечной ткани (то есть активность ферментной цепи переноса электронов) и с помощью анализа мышечной митохондриальной ДНК.[20]
Уровень молочной кислоты и пировиноградной кислоты крови, как правило, повышается в результате увеличения анаэробного метаболизма и уменьшением соотношения АТФ/АДФ. При исследовании СМЖ обнаруживается повышенный уровень белка, как правило, > 100 мг/дл, а также повышенный уровень молочной кислоты.[10]
В настоящее время нет медицинского лечения для KSS. Потому что это редкое состояние, есть только сообщения о случаях лечения с очень небольшим количеством данных, чтобы говорить об их эффективности. Было зарегистрировано несколько перспективных открытий, которые могут поддержать открытие новых методов лечения при дальнейших исследованиях. Спутниковые клетки отвечают за регенерацию мышечных волокон. Было отмечено, что мутант мтДНК не обнаруживается или редко обнаруживается в сателлитных клеток, культивируемых у пациентов с KSS. Shoubridge и др. (1997) задался вопросом, может ли первотип мтДНК быть восстановлен в мышечной ткани, при стимуляции регенерации мышц? В вышеупомянутом исследовании, регенерирующие мышечные волокна были отобраны в исходной биопсии, и было обнаружено, что они существенно гомоплазмичны для первотипа мтДНК.[16] Возможно, будущие методы содействия регенерации мышечных клеток и пролиферации спутниковых клеток, позволят значительно улучшить функциональное состояние KSS пациентов. Одно исследование описывало пациента с KSS, которое позволило сократить уровень сыворотки кофермента Q10. Введение 60-120 мг коэнзима Q10 в течение 3 месяцев привело к нормализации уровней молочной кислоты и пировиноградной кислоты, улучшению ранее диагностированной первой степени AV блока, и совершенствованию глазных движений.[21]
Скрининг ЭКГ рекомендуется для всех пациентов с CPEO. При KSS рекомендуется имплантация кардиостимулятора, следить за развитием значительного нарушения сердечной проводимости, даже у бессимптомных пациентов.[22]
Скрининг эндокринологических нарушений должен быть выполнен, включая измерение уровня глюкозы в сыворотке крови, функции щитовидной железы, уровней кальция и магния и уровней электролита в сыворотке. Гиперальдостеронизм наблюдается у 3 % пациентов с KSS.[23]
- ↑ Harvey J.N., Barnett D. Endocrine dysfunction in Kearns-Sayre syndrome (неопр.) // Clin. Endocrinol. (Oxf). — 1992. — July (т. 37, № 1). — С. 97—103. — doi:10.1111/j.1365-2265.1992.tb02289.x. — PMID 1424198.
- ↑ Kearns T.P., Sayre G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases (англ.) // AMA Arch Ophthalmol : journal. — 1958. — August (vol. 60, no. 2). — P. 280—289. — doi:10.1001/archopht.1958.00940080296016. — PMID 13558799.
- ↑ Jager B.V., Fred H.L., Butler R.B., Carnes W.H. Occurrence of retinal pigmentation, ophthalmoplegia, ataxia, deafness and heart block. Report of a case, with findings at autopsy (англ.) // The American Journal of Medicine (англ.)русск. : journal. — 1960. — November (vol. 29, no. 5). — P. 888—893. — doi:10.1016/0002-9343(60)90122-4. — PMID 13789175.
- ↑ 1 2 Kearns T.P. External Ophthalmoplegia, Pigmentary Degeneration of the Retina, and Cardiomyopathy: A Newly Recognized Syndrome (англ.) // Trans Am Ophthalmol Soc : journal. — 1965. — Vol. 63. — P. 559—625. — PMID 16693635.
- ↑ Zeviani M., Moraes C.T., DiMauro S., et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome (англ.) // Neurology (англ.)русск. : journal. — Wolters Kluwer (англ.)русск., 1988. — September (vol. 38, no. 9). — P. 1339—1346. — doi:10.1212/wnl.38.9.1339. — PMID 3412580.
- ↑ Lestienne P., Ponsot G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion (англ.) // The Lancet : journal. — Elsevier, 1988. — April (vol. 1, no. 8590). — P. 885. — doi:10.1016/S0140-6736(88)91632-7. — PMID 2895391.
- ↑ Carod-Artal F.J., Lopez Gallardo E., Solano A., Dahmani Y., Herrero M.D., Montoya J. [Mitochondrial DNA deletions in Kearns-Sayre syndrome] (исп.) // Neurologia. — 2006. — Сентябрь (т. 21, № 7). — С. 357—364. — PMID 16977556.
- ↑ Lertrit P., Imsumran A., Karnkirawattana P., et al. A unique 3.5-kb deletion of the mitochondrial genome in Thai patients with Kearns-Sayre syndrome (англ.) // Human Genetics : journal. — 1999. — Vol. 105, no. 1—2. — P. 127—131. — doi:10.1007/s004390051074. — PMID 10480366. Архивировано 29 сентября 2000 года.
- ↑ Soga F., Ueno S., Yorifuji S. [Deletions of mitochondrial DNA in Kearns-Sayre syndrome] (яп.) // Nippon Rinsho. — 1993. — Сентябрь (т. 51, № 9). — С. 2386—2390. — PMID 8411717.
- ↑ 1 2 Syndrome 950897, раздел Kearns-Sayre Syndrome (англ.) на сайте EMedicine
- ↑ 1 2 Walsh & Hoyt's Clinical Neuro-Ophthalmology: The Essentials (англ.) / Miller, Neil R.; Newman, Nancy J.. — Lippincott Williams & Wilkins (англ.)русск., 2007.
- ↑ Garcia-Cazorla A., Quadros E.V., Nascimento A., Garcia-Silva M.T., Briones P., Montoya J et al. Mitochondrial diseases associated with cerebral folate deficiency (англ.) // Neurology (англ.)русск. : journal. — Wolters Kluwer (англ.)русск., 2008. — Vol. 70, no. 16. — P. 1360—1362. — doi:10.1212/01.wnl.0000309223.98616.e4. — PMID 18413591.
- ↑ Quijada-Fraile P., O'Callaghan M., Martín-Hernández E., Montero R., Garcia-Cazorla À, de Aragón AM et al. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome (англ.) // Orphanet Journal of Rare Diseases (англ.)русск. : journal. — 2014. — Vol. 9. — P. 217. — doi:10.1186/s13023-014-0217-2. — PMID 25539952.
- ↑ Spector R., Johanson C.E. Choroid plexus failure in the Kearns-Sayre syndrome (англ.) // Cerebrospinal Fluid Res : journal. — 2010. — Vol. 7. — P. 14. — doi:10.1186/1743-8454-7-14. — PMID 20731822.
- ↑ Fine P.E. Mitochondrial inheritance and disease (англ.) // The Lancet. — Elsevier, 1978. — September (vol. 2, no. 8091). — P. 659—662. — doi:10.1016/S0140-6736(78)92764-2. — PMID 80581.
- ↑ 1 2 Shoubridge E.A., Johns T., Karpati G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy (англ.) // Human Molecular Genetics (англ.)русск. : journal. — Oxford University Press, 1997. — December (vol. 6, no. 13). — P. 2239—2242. — doi:10.1093/hmg/6.13.2239. — PMID 9361028.
- ↑ Fischel-Ghodsian N., Bohlman M.C., Prezant T.R., Graham J.M., Cederbaum S.D., Edwards M.J. Deletion in blood mitochondrial DNA in Kearns-Sayre syndrome (англ.) // Pediatric Research (англ.)русск. : journal. — 1992. — June (vol. 31, no. 6). — P. 557—560. — doi:10.1203/00006450-199206000-00004. — PMID 1635816.
- ↑ 1 2 Poulton J., Morten K.J., Weber K., Brown G.K., Bindoff L. Are duplications of mitochondrial DNA characteristic of Kearns-Sayre syndrome? (англ.) // Human Molecular Genetics (англ.)русск. : journal. — Oxford University Press, 1994. — June (vol. 3, no. 6). — P. 947—951. — doi:10.1093/hmg/3.6.947. — PMID 7951243.
- ↑ Miller, Neil R.; Newman, Nancy J.; Bioussee, Valerie; Kerrison, John B. Ch. 20, adapted from a chapter 22 by Paul N. Hoffman // Walsh and Hoyt's Clinical Neuro-ophthalmology: the essentials (англ.). — Philadelphia: Lippincott Williams & Wilkins (англ.)русск., 2008. — P. 432—436.
- ↑ Rubin, Richard M.; Sadun, Alfredo A. Ch. 9.17 Ocular Myopathies // Ophthalmology (неопр.) / Yanoff, Myron; Duker, Jason. — 3rd. — Mosby, 2008.
- ↑ Ogasahara S., Yorifuji S., Nishikawa Y., et al. Improvement of abnormal pyruvate metabolism and cardiac conduction defect with coenzyme Q10 in Kearns-Sayre syndrome (англ.) // Neurology (англ.)русск. : journal. — Wolters Kluwer (англ.)русск., 1985. — March (vol. 35, no. 3). — P. 372—377. — doi:10.1212/WNL.35.3.372. — PMID 3974895.
- ↑ Gregoratos G., Abrams J., Epstein A.E., et al. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/NASPE Committee to Update the 1998 Pacemaker Guidelines) (англ.) // Circulation (англ.)русск. : journal. — Lippincott Williams & Wilkins (англ.)русск., 2002. — October (vol. 106, no. 16). — P. 2145—2161. — doi:10.1161/01.CIR.0000035996.46455.09. — PMID 12379588.
- ↑ Syndrome 950897, раздел Kearns-Sayre Syndrome (англ.) на сайте EMedicine
Синдром Ади — Википедия
Материал из Википедии — свободной энциклопедии
Синдром Ади (Adie), который иногда называют синдромом Холмс-Ади или тоник зрачок Ади , — неврологическое расстройство, характеризующееся монотонно расширяющимся зрачком, который медленно реагирует на свет, но показывает более определенную реакцию аккомодации (то есть ярко-близкую диссоциацию).[1] Он часто наблюдается у женщин, у которых отсутствуют коленный или Ахиллов рефлексы. Если дополнительно нарушено потоотделение, его называют сидромом Росса (Ross). Он назван в честь британского невролога Уильяма Джон Ади.(англ. William John Adie) Синдром обусловлен повреждением постганглионарных волокон в парасимпатической иннервации глаза, как правило, вследствие вирусной или бактериальной инфекции, которая вызывает воспаление и влияет на зрачок глаза и вегетативную нервную систему.[1]
Синдром Ади представляет три отличительных признака, а именно, по меньшей мере один аномально расширенный зрачок (мидриаз), который не откликается на свет, потери глубоких сухожильных рефлексов и нарушения потоотделения.[1] Другие признаки могут включать в себя дальнозоркость вследствие аккомодативного пареза , светобоязнь и трудности при чтении.[2]
Зрачковые симптомы синдрома Холмс-Ади, как считается, результат вирусной или бактериальной инфекции, которая вызывает воспаление и повреждение нейронов в цилиарной ганглии, расположенной в задней части орбиты, обеспечивающей парасимпатический контроль глазного сужения. Кроме того, у пациентов с синдромом Холмс-Ади могут также возникнуть проблемы с вегетативным контролем тела. Этот второй набор симптомов вызван повреждением спинальных ганглиев в спинном мозге.[1]
Клинический тест может выявить секторальный парез сфинктера радужной оболочки или её червеобразное движение. Тоник зрачок может стать меньше (мейотического) за время, которое называют «little old Adie’s».[3][4] Тестирование с низкой дозой (1/8%) пилокарпина тоник зрачок может сжиматься из-за сверхчувствительной холинергической денервации.[1] Нормальный зрачок не будет сжиматься от разбавленной дозы пилокарпина.[4]КТ и МРТ сканирование может быть полезным в диагностическом тестировании координационных гипоактивных рефлексов.[5]
Обычное лечение стандартизированного синдрома Ади состоит в прописывании очков для чтения, корректирующих нарушений в глазу(ах).[1] Капли пилокарпина могут быть введены в качестве лечения, а также в качестве диагностического средства.[1] Грудная симпатэктомия является окончательным лечением потливости, если это не поддается лечению лекарственной терапией.[1]
Синдром Ади не угрожает жизни или трудоспособности.[1] Таким образом, нет рейтинга смертности для этого состояния; однако, потеря глубоких сухожильных рефлексов является постоянным и может прогрессировать с течением времени.[1]
Синдром Ади наиболее часто поражает молодых женщин (2,6: 1 с перевесом женщин) и является односторонним в 80 % случаев.[4] Средний возраст начала заболевания 32 года.
Синдром Аперта — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 декабря 2015; проверки требуют 12 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 декабря 2015; проверки требуют 12 правок.Синдром Апера (Аперта) (акрокраниодисфалангия, акросфеносиндактилия, акроцефалосиндактилия) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии[1]. Синдром Апера является одной из форм акроцефалосиндактилии[прим. 1].
Впервые это заболевание описал французский педиатр Эжен Аперт в 1906 году[2]. Заболевание встречается у одного новорожденного на 160 000—200 000[3].
Акрокраниодисфалангия — заболевание с аутосомно-доминантным типом наследования[прим. 2]. У детей, родители которых страдают синдромом Аперта, вероятность унаследовать заболевание равна 50%. В 1995 году были опубликованы доказательства, судя по которым, причиной возникновения данного синдрома является нарушение работы фибробластов генов 10-й хромосомы человека[4].
Синдактилия двух пальцев у младенцаОдним из признаков заболевания является то, что краниосиностоз[прим. 3] сочетается с брахикефалией. В связи с преждевременным срастанием коронарных швов увеличивается внутричерепное давление, что обычно приводит к умственной отсталости. Немаловажными признаками синдрома являются высокий, выпуклый лоб, плоское или вогнутое лицо, в результате чего наблюдается нарушение костей лицевого черепа, что приводит к деформации челюсти, а также синдактилия рук и ног с вовлечением 2, 3 и 4-го пальцев.
Радикальных методов лечения не существует.
Симптоматическое лечение данной патологии заключается в хирургическом увеличении объёма черепа, коррекция синдактилии и полидактилии.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Апера часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.[5]
Консервативные методы лечения применяют для разработки суставов. Также, для стимуляции психического развития назначают ноотропные препараты: аминалон, пирацетам и другие[1], использовались в эпоху развития науки до проверки лечения методами доказательной медицины.
- ↑ Акроцефалосиндактилия — группа наследственных пороков развития черепа и пальцев.
- ↑ Аутосомно-доминантный тип наследования — тип наследования, при котором мутантный аллель доминирует над нормальным аллелем, в связи с чем болезнь или признаки болезни ярко выражены.
- ↑ Краниосиностоз — преждевременное сращение некоторых костей черепа.
- ↑ 1 2 Л.О. Бадалян. Детская неврология. — Москва: Медицина, 1984. — С. 347—349. — 576 с.
- ↑ Apert's syndrome (whonamedit.com) (англ.). Дата обращения 13 августа 2010. Архивировано 6 мая 2012 года.
- ↑ Kaplan, L C. Clinical assessment and multispecialty management of Apert syndrome (англ.) // Clinics in plastic surgery : journal. — 1991. — April (vol. 18, no. 2). — P. 217—225. — ISSN 00941298. — PMID 2065483.
- ↑ Wilkie, A O; S. F. Slaney, M. Oldridge, M. D. Poole, G. J. Ashworth, A. D. Hockley, R. D. Hayward, D. J. David, L. J. Pulleyn, P. Rutland. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome (англ.) // Nature genetics : journal. — 1995. — February (vol. 9, no. 2). — P. 165—172. — doi:10.1038/ng0295-165. — PMID 7719344.
- ↑ Синдром Апера: клинические проявления и этиология — Интернет-сообщество нейрохирургов Росcии (неопр.). neuro-online.ru. Дата обращения 4 марта 2017.
Синдром Коккейна — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 3 декабря 2015; проверки требуют 9 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 3 декабря 2015; проверки требуют 9 правок.Синдром Коккейна (англ. Cockayne syndrome, CS), также называемый синдром Нил-Дингуолл (англ. Neill-Dingwall)) — редкое аутосомно-рецессивное[2], нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением[3][4]. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты[4]. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия (состояние, характеризующееся деградацией белого вещества). В основе расстройства лежит дефект механизма репарации ДНК[5]. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции[6]. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы[6].
Синдром назван в честь английского врача Эдварда Альфреда Коккейна (англ. Edward Alfred Cockayne)(1880-1956), который первым описал его в 1936 году и повторно в 1946 году в статье под названием «Карликовость с атрофией сетчатки и глухота (англ. Dwarfism with Retinal Atrophy and Deafness) »[7]. Синдром Нил-Дингуолл был назван в честь Марии М. Дингуолл (англ. Mary M. Dingwall) и Кэтрин А. Нилл (англ. Catherine A. Neill)[7]. Эти женщины описали случай двух братьев с синдромом Коккейна и утверждали, что это та же болезнь, что описана Эдвардом Альфредом Коккейном. В своей статье женщины объясняли симптомы заболевания в связи с их открытием кальцификации головного мозга. Они также сравнили синдром Коккейна с тем, что сейчас известно как синдром Хатчинсона-Гилфорда (HGPS) под названием Прогерия, в связи с прогрессированием старения, которое характеризует оба расстройства[7].
- CS I типа, «классический» вид, характеризуется нормальным ростом плода с наступлением аномалии в течение первых двух лет жизни. Зрение и слух постепенно снижаются[8]. Центральная и периферическая нервные системы постепенно вырождаются до самой смерти в первом или втором десятилетии жизни, в результате серьезного неврологического ухудшения. Атрофия коры головного мозга менее серьезна в CS типа I[9].
- CS II типа присутствует с рождения (врожденная) и гораздо более серьезная, чем CS типа 1[8]. Это предполагает очень слабое неврологическое развитие после рождения. Смерть обычно наступает в семилетнем возрасте. Этот тип также обозначается как церебральный окуло-фацио-скелетный (COFS) синдром или синдром Пенья-Шокейр (англ. Pena-Shokeir) типа II[8]. Синдром COFS назван так из-за последствий, которые он оказывает на мозг, глаза, лицо и скелетную систему, а болезнь часто приводит к атрофии головного мозга, катаракте, потере жирности лица и остеопорозу. Синдромы COFS могут быть дополнительно подразделены на несколько условий (типа COFS 1, 2, 3 (связанные с пигментной ксеродермой) и 4)[10]. Обычно пациенты с этим видом рано начавшегося расстройства демонстрируют также серьезные повреждения головного мозга, в том числе снижению миелинизации белого вещества и более широко распространенную кальцификацию, в том числе в коре головного мозга и базальных ганглиев[9].
- CS типа III, характеризуются поздним началом, течение как правило, мягче, чем у типов I и II[8]. Часто пациенты с типом III доживают до взрослого возраста.
- Пигментная Ксеродерма-синдром Коккейн (XP-CS) происходит, когда человек попутно страдает от пигментной ксеродермы, другого заболевания репарации ДНК. Экспрессируются некоторые симптомы каждой болезни. Например, присутствуют веснушки и пигментные нарушения, характерные XP. Заметны характерные CS неврологические расстройства, спастичность и неразвитость половых органов. Тем не менее, отсутствуют гипомиелинизация и типичные черты лица пациентов CS[11].
Лица с этим синдромом имеют меньший обычного размер головы (микроцефалия), невысокий рост (карликовость), глаза выглядят запавшими, и они имеют «пожилой» вид. Они часто имеют длинные конечности с контрактурами суставов (неспособностью расслабления мышцы в суставе), сутулость (кифоз) и они могут быть очень тощими (кахексия) из-за потери подкожного жира. Их маленький подбородок, большие уши, и заостренный, тонкий нос часто дают пожилой внешний вид[9]. Кожа лиц с синдромом Коккейна также часто аномальна. Гиперпигментация, варикозные или узловатые вены (телеангиэктазия)[9] и серьезная чувствительность к солнечному свету являются общими симптомами, даже у лиц без XP-CS. Часто пациенты с синдромом Коккейн бывают слабо чувствительны к ожогам и волдырям. Глаза пациентов могут быть затронуты различными путями и их аномалии распространены в CS. Катаракта и помутнение роговицы являются общими симптомами. Вполне может произойти потеря и повреждение элементов зрительного нерва, вызывающая его атрофию[4]. Нистагм, или непроизвольные движения глаз и зрачки, которые не расширяются, показывают потерю управления и непроизвольного сокращения мышц[9]. Соль и перец пигментации сетчатки также очевидный симптом. Диагноз определяется конкретным тестом для репарации ДНК, который измеряет восстановление РНК после воздействия УФ-излучения.
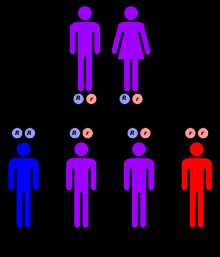 Синдром Коккейн имеет аутосомно-рецессивный характер наследования.
Синдром Коккейн имеет аутосомно-рецессивный характер наследования. Синдром Коккейн классифицируется генетически следующим образом:
Мутации в гене ERCC6 (также известном как ген CSB) или ERCC8 (также известном как ген CSA) являются причиной синдрома Коккейн[8]. Белки от этих генов, участвующих в репарации ДНК с помощью транскрипционной связи механизма репарации, в частности ДНК в активных генах. Повреждение ДНК, вызываются ультрафиолетовыми лучами от солнечного света, радиацией или свободными радикалами в организме. Нормальная клетка может восстановить повреждения ДНК легко, прежде чем они накопятся. Если какой-либо ген ERCC6 или ERCC8 изменяется (как в синдроме Коккейн), повреждение ДНК не устраняются. По мере накапления повреждений, это может привести к неполноценным клеткам или к гибели клеток. Это гибель клеток и их неполноценность, вероятно, способствует симптомам синдрома Коккейн, таким как преждевременное старение и гипомиелинизация нейронов[8].
Мутации в генной мутации ERCC6 составляет ~ 70% случаев.
Визуальные исследования показывают широкое отсутствие миелиновых оболочек нейронов в белом веществе головного мозга, и общую атрофию коры головного мозга[6]. Кальцификаты также были найдены в скорлупе, площади переднего мозга, которая регулирует движения и способствует некоторым формам обучения[9], вместе с корой[7]. Кроме того, атрофия центральной части мозжечка у больных с синдромом Коккейна также может привести к отсутствию мышечного управления, в частности, к обычно наблюдаемой непроизвольной и плохой осанке.
Не существует постоянного лечения этого синдрома, хотя пациенты могут лечиться в соответствии с их специфическими симптомами. Прогноз для больных синдромом Коккейна плохой, а смерть, как правило, происходит у каждой двадцатой личности. Лечение обычно включает в себя физиотерапию и незначительные операции на пораженных органах, такие как удаление катаракты[4]. Кроме того, рекомендуется также наносить толстый слой солнцезащитного крема и носить защитную одежду пациентам с синдромом Коккейна, слишком чувствительным к УФ-излучению[12]. Может также помочь оптимальное питание. Для родителей рекомендуется генетическая консультация, так как расстройство имеет 25% шанс передачи любым будущим детям, и также по возможности рекомендуется пренатальное тестирование[4].
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Bertola, Dr; Cao, H; Albano, Lm; Oliveira, Dp; Kok, F; Marques-Dias, Mj; Kim, Ca; Hegele, Ra. Cockayne syndrome type A: novel mutations in eight typical patients (англ.) // Journal of Human Genetics (англ.)русск. : journal. — 2006. — Vol. 51, no. 8. — P. 701—705. — doi:10.1007/s10038-006-0011-7. — PMID 16865293.
- ↑ James, William; Berger, Timothy; Elston, Dirk. Andrews' Diseases of the Skin: Clinical Dermatology (англ.). — 10th (англ.)русск.. — Saunders, 2005. — P. 575. — ISBN 0-7216-2921-0.
- ↑ 1 2 3 4 5 Bender M, Potocki L, Metry D. What syndrome is this? Cockayne syndrome. Pediatric Dermatology [serial online]. November 2003;20(6):538-540. Available from: MEDLINE with Full Text, Ipswich, MA. Accessed April 30, 2015.
- ↑ Hoeijmakers J.H. DNA damage, aging, and cancer (англ.) // The New England Journal of Medicine. — 2009. — October (vol. 361, no. 15). — P. 1475—1485. — doi:10.1056/NEJMra0804615. — PMID 19812404.
- ↑ 1 2 3 Nance M, Berry S. Cockayne syndrome: review of 140 cases. American Journal Of Medical Genetics [serial online]. January 1, 1992;42(1):68-84. Available from: MEDLINE with Full Text, Ipswich, MA. Accessed April 30, 2015.
- ↑ 1 2 3 4 Neill CA, Dingwall MM. A Syndrome Resembling Progeria: A Review of Two Cases. Archives of Disease in Childhood. 1950;25(123):213-223.
- ↑ 1 2 3 4 5 6 Cockayne Syndrome. Genetics Home Reference http://ghr.nlm.nih.gov/condition/cockayne-syndrome Published April 28, 2015. Reviewed May 2010. Accessed April 30, 2015.
- ↑ 1 2 3 4 5 6 Javadzadeh M. Cockayne Syndrome. Iran J Child Neurol. Autumn 2014;8;4(Suppl.1):18-19.
- ↑ Cerebrooculofacioskeletal Syndrome 2. Online Mendelian Inheritance in Man. https://omim.org/entry/610756. Published 2/12/2007.
- ↑ Laugel V. Cockayne Syndrome. 2000 Dec 28 [Updated 2012 Jun 14]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: [1]
- ↑ Kyllermen, Marten. Cockayne Syndrome. Swedish Information Centre for Rare Diseases. 2012: 4.0. http://www.socialstyrelsen.se/rarediseases/cockaynesyndrome#anchor_17 Архивная копия от 24 сентября 2015 на Wayback Machine