Синдром крузона что это такое
Синдром Крузона: причины, признаки, лечение, прогноз
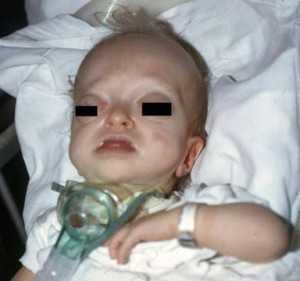
Синдром Крузона – деформация лицевой и мозговой части черепа, формирующаяся в период внутриутробного развития и передающаяся по наследству. Патология возникает достаточно редко, протекает тяжело и сопровождается аномальным преждевременным заращением швов между черепными и лицевыми костями. Раннее закрытие черепных швов способствует ограниченному объему черепа и изменению формы головы. У больных нос приобретает крючковидную форму, нарушается слух и зрение. При поражении коронарного шва, идущего от одного уха к другому, возникают аномалии глазниц – они становятся выпуклыми. У больных изменяется высота лба и выраженность челюстной мускулатуры. Косметические уродства часто сочетаются с задержкой интеллекта или умственной отсталостью.
Краниофасциальный дизостоз характеризуется нарушением процессов окостенения черепа. Это заболевание впервые описал педиатр из Франции Крузон в 1912 году. С тех пор оно носит имя своего первооткрывателя. Патологическое состояние одинаково часто встречается как среди мальчиков, так и среди девочек. Точная частота возникновения данного недуга в настоящее время остается неизвестной.
первый пациент с описанным синдромом Крузона (1912 г.)
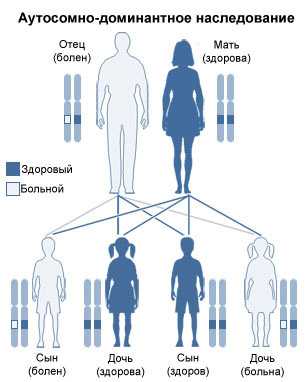
Синдром Крузона наследуется по аутосомно-доминантному механизму. В некоторых случаях причиной патологии может стать спонтанная мутация генов. У новорожденных детей имеются швы между костями лица и черепа. По мере развития детского организма череп расширяется благодаря этим швам. У здоровых людей они полностью зарастают лишь после остановки роста мозга и его полноценного формирования. При синдроме Крузона черепные кости срастаются намного раньше. Череп больных растет в сторону открытых швов, что приводит к появлению выраженных дефектов внешности.
Нередко деформация черепа сочетается с аномалиями отдельных структур опорно-двигательного аппарата — межпозвоночных сочленений, крупных суставов конечностей: локтевого, плечевого, коленного. Краниосиностоз осложняется развитием гидроцефалии — водянки головного мозга, приводящей к повышению внутричерепного давления. Тяжелая деформация лицевых костей сопровождается нарушением носового дыхания. Особенно опасно подобное расстройство для новорожденных детей, которые еще не умеют дышать ртом. Возможно появление ночного апноэ – внезапной остановки дыхания во сне.
Диагностика синдрома заключается в тщательном визуальном осмотре больного, проведении рентгенологического и молекулярно-генетического исследований. Синдром Крузона — неизлечимое заболевание. Для поддержания жизни больных на оптимальном уровне проводят симптоматическую терапию консервативного и хирургического характера. Краниопластика и другие корригирующие оперативные вмешательства устраняют деформации черепа.
Причины
Синдром Крузона — наследственная патология, возникающая вследствие мутационного процесса. Передается недуг по аутосомно-доминантному принципу от родителей детям. Когда в одной родительской хромосоме присутствует мутировавший ген, дети не всегда наследуют заболевание. В таких семьях риск рождения ребенка с синдромом Крузона составляет 50%. Причем новорожденные дети могут даже не быть носителями дефекта. Тщательное обследование на этапе планирования беременности позволяет повысить шанс рождения здорового малыша.
Данный недуг развивается при мутации гена, расположенной в X хромосоме. Этот ген является нестабильным из-за значительных размеров и большого количества экзонов. Его мутации приводят к развитию наследственных патологий скелета человека. Происходит нарушение структуры соединительной ткани, костей и хрящей. В области межкостных швов скапливаются фибробласты, синтезирующие межклеточное вещество, продуцирующие белки — коллаген и эластин и активизирующие процессы окостенения. Это основной этиопатогенетический фактор синдрома Крузона.
К факторам, способствующим развитию патологии, относятся: наличие патологии в семейном анамнезе, возраст отца старше 60 лет в период зачатия ребенка.
Известны случаи ненаследственного характера синдрома Крузона, развивающегося на фоне спонтанной мутации в гене. Чаще всего такие нарушения встречаются по отцовской линии.
Симптоматика
Клинические признаки синдрома Крузона весьма специфичны. Они становятся заметными сразу после рождения. Выраженность проявлений достигает своего максимума ближе к 3-4 годам.
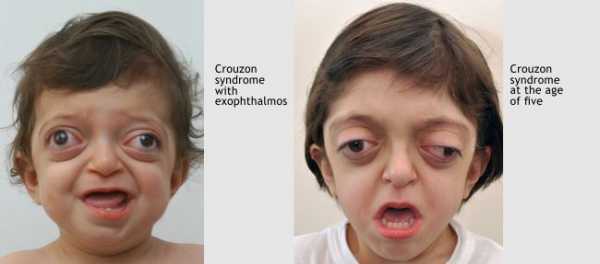
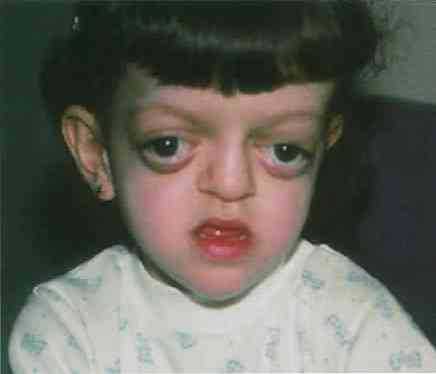
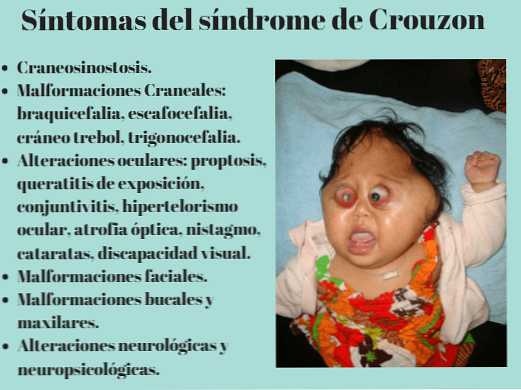
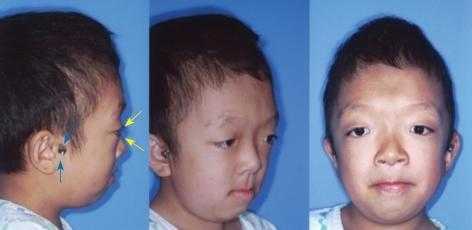
- Основной симптом патологии – краниосиностоз, при котором прочно срастаются кости черепа по венечному или стреловидному шву. При этом рост головы останавливается, и ребенок приобретает характерные черты: дистальный прикус, большое расстояние между глазами, клювовидный нос, низкое расположение ушей, выступающий язык, укороченная и низко расположенная губа, недостаточное смыкание челюстей, редкие зубы, высокое небо, выдающийся вперед подбородок, расщелина на язычке. Характерная черта синдрома — неполное смыкание зубов.
- Дополнительные признаки – синдактилия пальцев, атрезия хоан, сколиоз, лордоз, мигрень, акантоз.
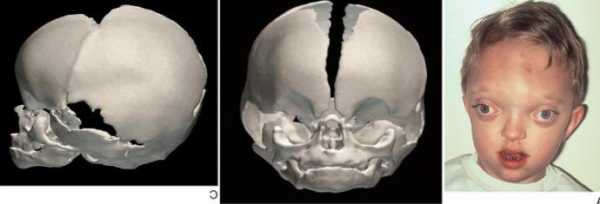
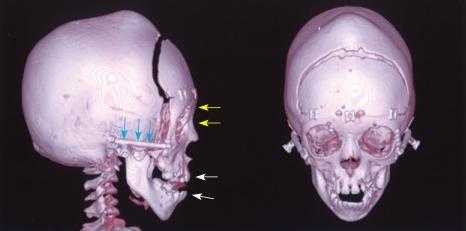
- По мере прогрессирования синдрома формируется короткоголовость – брахицефалия. Возможные виды деформации черепной коробки: клиновидная или ладьевидная голова, гидроцефалоидная деформация черепа в форме трилистника. При пальпации определяются уплощенные края швов.
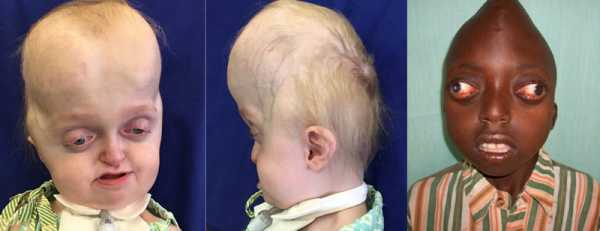
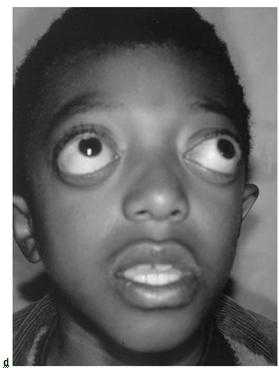
- Поражение зрительного анализатора проявляется расходящимся косоглазием, нистагмом, эктопией, колобомой. Экзофтальм обусловлен незначительной глубиной орбит. У больных раскосые глаза постоянно вращаются, совершая спонтанные движения. Значительно снижается острота зрения. У 80% больных выявляют частичную атрофию зрительного нерва.
- Нарушения слуха вплоть до полной глухоты происходит при поражении пирамиды височной кости, фиксации и деформации слуховых косточек. У больных изменяется форма внутреннего слухового канала, понижается звуковая проводимость костей, развивается атрезия внешнего слухового хода.
- Поражение нервной системы – умственная отсталость, задержка речи, судороги. Нарушения интеллекта сохраняются на протяжении всей жизни, затрудняя межличностное общение в социуме.
Синдром Крузона — серьезный недуг, приводящий к развитию у ребенка различных осложнений.
Негативные последствия заболевания:
- гидроцефалия — водянка мозга,
- потеря зрения — результат сдавливания зрительного нерва и его атрофии,
- язвенный кератит развивается при неполном смыкании век и частичном высыхании роговицы,
- умственная неполноценность,
- трудности с адаптацией в обществе.
Диагностические мероприятия
Диагностика патологии включает следующие методики:
- общий осмотр позволяет обнаружить характерные признаки синдрома;
- сбор семейного анамнеза проводится с целью выявления случаев заболевания в роду;
- рентгенологическое исследование — оценка формы черепа и наличия сращений швов, расширение ямок гипофиза, уплощение глазницы со своеобразными вдавлениями;
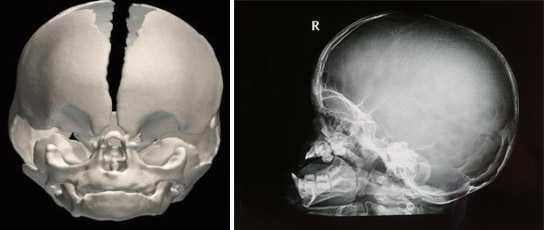
- ядерно-резонансная или компьютерная томография — гиперостозы, аномалии слуховых косточек, отсутствие полости среднего уха, опущение миндалин мозжечка;
- генетическое обследование и молекулярная диагностика методом ПЦР — выявление мутации в 7 и 9 экзонах гена десятой хромосомы;
- осмотр у офтальмолога, определение остроты зрения, рефрактометрия;
- консультация ЛОР-врача и аудиометрия;
- работа с психотерапевтом или неврологом, позволяющая определить уровень психического развития больного.
Лечение
Синдром Крузона — неизлечимое заболевание, требующее функциональной и косметической коррекции. Достичь ее можно лишь оперативным путем.
Хирургическое вмешательство направлено на восстановление формы черепа и устранение синостозов. Такие операции проводят, начиная с рождения и до остановки роста черепной коробки. Они позволяют снизить уровень внутричерепного давления, исправить костно-мышечные деформации и предотвратить дальнейшее прогрессирование умственной отсталости, а также психоневрологических нарушений. Для этого раскрывают рано заращенные швы черепа и исправляют форму лица.
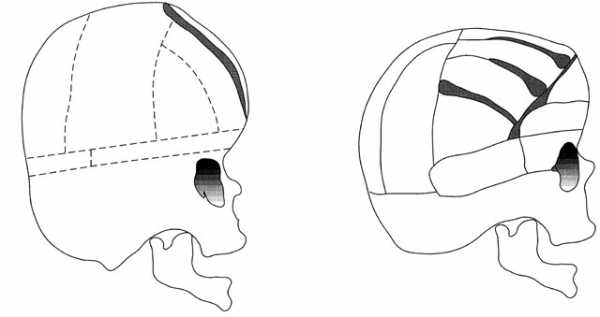
Основные виды операций:
- Краниопластика и вскрытие синостозированных швов – удаление и расширение фрагментов костей черепа для предотвращения повреждение мозга и исправления формы черепной коробки.
- Создание искусственного блефарофимоза для снижения выраженности экзофтальма и исправления положения глазного яблока.
- Расширение хоан для восстановления дыхания.
- Установка вентрикулоперитонеальной шунтирующей системы при гидроцефалии.
- Использование аппарата Илизарова для выдвижения вперед верхней челюсти и улучшения дыхательной функции.
- Коррекция выступающей нижней челюсти и нормализация ее внешнего вида.
- Радикальные комплексные операции направлены на устранение дефектов лица – исправление верхнечелюстной гипоплазии, восстановление зубного ряда.
В настоящее время внедряются в хирургическую практику дистракционные методики коррекции деформаций лицевых костей. С помощью специальных аппаратов для смещения любого участка черепа и исправления костных дефектов процесс лечения проходит быстрее и более эффективно.
 постоперационные приспособления для улучшения эффективности лечения
постоперационные приспособления для улучшения эффективности лечения В тех случаях, когда операция противопоказана ребенку, нарушение функции дыхания компенсируют постоянной кислородотерапией, дыханием под повышенным давлением, использованием воздуховодов, трахеостомией.
Облегчить состояние больного также поможет лекарственная терапия. Больным назначают:
- Ноотропы – «Пантогам», «Пирацетам», «Винпоцетин».
- Сосудистые препараты – «Кавинтон», «Циннаризин», «Актовегин».
- Диуретики – «Лазикс», «Верошпирон», «Диакарб».
Больные с синдромом Крузона находятся на учете у офтальмологов и оториноларингологов, которые при выявлении каких-либо отклонений назначают адекватную терапию. Дети с психическими расстройствами нуждаются в специальном лечении и занятиях с логопедом.
Прогноз
Прогноз при синдроме Крузона неоднозначный. Он зависит от выраженности черепной деформации и состояния мозговой ткани. Первоначальная реконструктивная операция проводится на первом году жизни. По мере роста ребенка она дополняется новыми вмешательствами на костях и мягких тканях лица.

Болезнь часто сопровождается задержкой психомоторного развития и судорожными приступами. Поэтому многие специалисты оценивают прогноз как неблагоприятный. Даже при проведении поддерживающей терапии в полном объеме у больных прогрессирует расходящееся косоглазие, развивается глухота и гипоплазия лица. Больные, вовремя обратившиеся за медицинской помощью и постоянно наблюдающиеся у специалистов, часто доживают до преклонных лет. Некоторые лица с синдромом Крузона все же сохраняют социальную адаптацию, несмотря на грубые косметические недостатки.
Причинами инвалидности являются нарушения зрения и слуха, а также умственная отсталость. Со временем дефекты костей становятся все более выраженными. Поскольку в настоящее время не разработаны профилактические мероприятия, предупреждающие развитие данного недуга, надеяться можно только на прогрессирование медицинских технологий, которые когда-нибудь смогут избавить людей от генетических расстройств.
Видео: мини-лекция о синдроме Крузона
Видео: фильм о синдроме Крузона
основные причины возникновения, признаки и варианты лечения
Синдром Крузона – редкая наследственная патология. Она сопряжена с формированием хромосомных нарушений, приводящих к сбоям в процессе развития костных структур черепа. Дети с данным заболеванием страдают от краниофациального дизостоза – сращения швов. Патология впервые описана Октавом Крузоном, в честь него недуг и получил свое название. Проблема сопровождается также нарушением слуха, зрения, выраженными косметическими дефектами, умственной отсталостью. Лечение заболевания паллиативное. Оно подразумевает как осуществление хирургического вмешательства, так и использование медикаментозных средств.
Причины возникновения синдрома Крузона
Патология относится к числу наследственных генетических проблем. При этом в медицине принято различать два варианта заболевания. Первый, классический, формируется вследствие возникновения дефекта в 10-й хромосоме. Участок ДНК данной локализации кодирует белок, восприимчивый к фактору роста фибробластов типа 2. Изменение структуры этого соединения сопровождается нарушением процессов формирования хрящевой и костной тканей черепа. Именно данный каскад реакций и обуславливает развитие специфической клинической картины. Причиной возникновения изменений при синдроме Крузона является преждевременное заращение швов в лицевой и мозговой частях черепа. Это происходит вследствие накопления в соединениях фибробластов, а затем несвоевременного запуска процессов окостенения. В норме синостоз отмечается лишь после завершения роста человеческого организма. При синдроме Крузона швы зарастают уже к 3–5 годам.
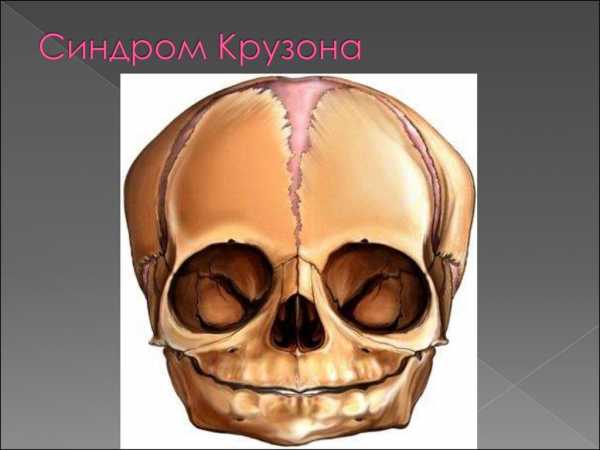
Второй вариант патологии сочетается с формированием кожных проявлений недуга. Он сопряжен с возникновением дефекта в 4-й хромосоме. Этот участок кодирует рецептор, восприимчивый к фактору роста фибробластов 3-го типа. Медики отмечают, что данный вид заболевания протекает тяжелее. Он носит название синдрома Октава Крузона с черным акантозом. У пациентов диагностируется краниофациальный дизостоз, сопровождающийся гиперкератозом, а также наличием большого количества родинок.
Обоим вариантам болезни свойственен аутосомно-доминантный тип наследования. В ряде случаев регистрируется возникновение спонтанных мутаций.
Характерные признаки
Клиническая картина генетического дефекта является специфической. Поэтому диагностика расстройства, как правило, не вызывает затруднений. К характерным симптомам синдрома Крузона относят:
- Краниосиностоз, то есть сращение швов лицевой и мозговой частей черепа. Подобный процесс сопровождается деформацией головы пациента. Это связано с тем, что в детстве происходит активный рост различных структур, в том числе и церебральных. В норме данный процесс сопровождается увеличением черепной коробки, что возможно, благодаря сохранению подвижного соединения между ее элементами. Синдром Крузона, провоцирующий сращение костей, препятствует нормальному развитию организма. Растущий головной мозг оказывает значительное давление на скелет, что провоцирует его деформацию. У детей отмечается уменьшение размеров челюстей, изменение нормального прикуса. Увеличивается расстояние между глазами, уши расположены ниже своей естественной локализации. У большинства пациентов развивается экзофтальм. По мере прогрессирования недуга при отсутствии лечения возможно выпадение глазных яблок из орбит.
- На фоне нарушения строения черепа и косметических дефектов присутствуют и другие симптомы. Дети с синдромом Крузона страдают от нарушений зрения. Развивается косоглазие, склонное к прогрессированию. Отмечается нистагм различной интенсивности, эктопия. При отсутствии лечения пациент теряет способность видеть.
- Из-за изменения строения черепа страдает и слуховой анализатор. Это связано с поражениями височных долей головного мозга, а также соответствующих костей. В ряде случаев у детей отсутствует барабанная перепонка. Подобные изменения зачастую приводят к полной глухоте.
- Синдром Октава Крузона сочетается с другими врожденными аномалиями. Среди них сращение пальцев на руках и ногах, атрезия хоан (зарастание носовых ходов), различные деформации позвоночника.
- Повышенное внутричерепное давление сопровождается неврологической симптоматикой. Дети отстают в умственном развитии, страдают от сильных мигреней и речевых дисфункций. При нарушении тока ликвора отмечается возникновение гидроцефалии. В тяжелых случаях у пациентов возникают судороги.
Согласно имеющимся исследованиям, синдром Крузона наследуется по аутосомно-доминантному типу. Расстройство характеризуется полной пенетрантностью и переменной экспрессией. При этом около трети всех случаев диагностики заболевания являются спонтанными мутациями.
В генетике описано около 30 различных дефектов хромосомного набора, которые приводят к возникновению специфической клинической картины.
При этом важно дифференцировать патологию от других проблем со сходными симптомами, в числе которых синдромы Апера, Карпентера и Пфайфера.
Интенсивность клинических проявлений поражения значительно варьируется. У некоторых детей отмечается легкое изменение строения черепа, в то время как другие страдают от стремительного и стойкого заращения краниальных швов и выраженного неврологического дефицита. Частым последствием этих изменений является обструкция дыхательных путей, происходящая на фоне атрезии хоан. Подобный дефект опасен тем, что сопровождается респираторным дистрессом, негативно сказывающимся на здоровье ребенка. Увеличение внутричерепного давления приводит к компрессии зрительного нерва. Данный каскад реакций обуславливает второе по распространенности осложнение синдрома Крузона – офтальмологические нарушения.
Диагностические исследования
Для подтверждения наличия болезни потребуется осмотр педиатра. Как правило, симптомы проявляются в возрасте 3–4 лет, когда происходит активное увеличение головного мозга в объеме. Пациенты имеют характерный внешний вид (представлены на фото), что позволяет докторам заподозрить наличие синдрома Крузона. Выявление в семейном анамнезе ребенка случаев диагностики генетического дефекта также свидетельствует о формировании данного нарушения. Информативными методами подтверждения проблемы являются рентген, компьютерная и магнитно-резонансная томография. Эти техники позволяют исследовать костные структуры и головной мозг. При этом точный диагноз подтверждается только на основании результатов генетического анализа. Для подготовки к дальнейшему лечению, подразумевающему проведение паллиативных операций, потребуется осуществление комплексного обследования. Оно включает в себя анализы крови, а также УЗИ. Проведение этих процедур имеет значение и для установления дальнейшего прогноза заболевания.
Лечение
Тактику борьбы с патологией определяет врач на основании результатов проведенного обследования. Методов, позволяющих полностью избавиться от аномалии, нет. Лечение носит симптоматический характер. При этом чем раньше пациенту будет оказана квалифицированная медицинская помощь, тем лучше дальнейший прогноз заболевания. Предпочтительно использование операционных техник, которые направлены на устранение синостозов и восстановление нормальной формы черепа. В ряде случаев используются и консервативные методы, направленные на коррекцию неврологического статуса пациента.
Хирургическое вмешательство
Основные риски при синдроме Крузона связаны с увеличением внутричерепного давления. В ходе операции устраняется неподвижное соединение краниальных швов, что обеспечивает возможность дальнейшего роста головного мозга без последствий для организма. Трудность заключается в том, что одного хирургического вмешательства, как правило, оказывается недостаточно. Они проводятся поэтапно, по мере развития ребенка. Кроме коррекции синостоза, применяются и другие техники:
- Искусственное сужение и укорочение глазной щели. Это необходимо для лечения экзофтальма и профилактики проптоза.
- Расширение хоан, направленное на восстановление естественного дыхания.
- При нарушении оттока ликвора и формировании гидроцефалии проводится шунтирование. Операция заключается в установлении специфической дренирующей системы, способствующей снижению внутричерепного давления.
- Различные косметические техники, направленные на восстановление нормальной формы челюсти, носа и зубного ряда.
Лекарственная терапия
Медикаментозная коррекция состояния пациента также используется. Однако в случае синдрома Крузона она имеет вспомогательное значение. Детям с генетической аномалией назначаются ноотропные средства, например, «Луцетам». Они способствуют поддержанию нормальной работы головного мозга. При повышении внутричерепного давления оправдано использование мочегонных препаратов, таких как «Фуросемид». В тяжелых случаях, сопровождающихся развитием судорог, применяются противоэпилептические медикаменты и транквилизаторы, например, «Диазепам». Большинству пациентов в период реабилитации после хирургического вмешательства выписываются нестероидные противовоспалительные препараты, обладающие анальгетическим действием, к которым относятся «Пенталгин» и «Диклофенак».
Опасность заболевания и прогноз
Отсутствие лечения может крайне неблагоприятно сказаться на состоянии здоровья. При увеличении объема головного мозга на фоне остановки роста черепа происходят фатальные изменения нервной ткани. Подобное состояние опасно поражением жизненно важных центров ЦНС и гибелью пациента. К числу распространенных осложнений относится также инвалидность вследствие потери слуха и зрения. Трудность при синдроме Крузона заключается и в социализации, поскольку еще в детском возрасте зачастую формируется умственная отсталость, препятствующая нормальному взаимодействию ребенка с окружающими. В таких случаях важную роль играет поддержка семьи.
Прогноз при заболевании неоднозначен. Своевременное и адекватное лечение способствует уменьшению интенсивности клинических проявлений недуга. При этом даже проведение хирургического вмешательства не обеспечивает полного выздоровления.
Профилактика
Предупреждение развития синдрома Крузона основано на генетическом анализе хромосомного набора будущих родителей. Важно и исключение негативного воздействия на плод в период беременности. Специфические средства профилактики патологии не разработаны.
Отзывы
Мария, 34 года, г. Ростов-на-Дону
Сын родился с синдромом Крузона. При этом никто в моей семье и в семье мужа не страдал от этой проблемы. У мальчика слишком рано заросли швы в черепе, из-за чего изменилась форма головы и лица. Ему пришлось провести несколько операций, чтобы снизить негативное воздействие на мозг. Лечение еще не окончено, но результаты уже внушительные.
Александр, 26 лет, г. Тюмень
У меня синдром Крузона. Это генетическое заболевание, при котором череп слишком рано затвердевает. Из-за этого в детстве у меня начало деформироваться лицо и голова. Родители возили по хирургам, пришлось сделать четыре операции, чтобы скорректировать изменения. Конечно, внешность осталась специфической, но осложнений удалось избежать.
Загрузка...Синдром Крузона - причины, симптомы, диагностика и лечение
Синдром Крузона – редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Общие сведения
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Синдром Крузона
Причины синдрома Крузона
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы синдрома Крузона
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Синдром Крузона с черным акантозом характеризуется аналогичными изменениями со стороны лица и черепа. При этом некоторые исследователи отмечают, что данная форма заболевания протекает в целом тяжелее и характеризуется повышенной частотой осложнений. Так, атрезия хоан, довольно редко развивающаяся при классической разновидности краниофасциального дизостоза 1 типа, в случае синдрома Крузона с черным акантозом регистрируется почти у половины больных. Кроме того, у пациентов наблюдаются сильно выраженные кожные нарушения – гиперкератоз (разрастание бородавок, гипертрофия кожи), усиленная пигментация. Основная локализация кожных проявлений при синдроме Крузона с черным акантозом – области коленных и локтевых сгибов, шея, живот, носогубные складки, зона вокруг глаз. Также для этого заболевании характерно наличие большого количества невусов (родинок), часто развиваются гипертрофические слабо пигментированные рубцы и шрамы.
Диагностика синдрома Крузона
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение синдрома Крузона
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз и профилактика синдрома Крузона
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Синдром крузона
Генетические заболевания в последнее время становятся чуть ли не основной проблемой современного мира. По данным ВОЗ 6% детей на Земном шаре страдают наследственными заболеваниями. В данной статье приводится случай синдрома Крузона у девочки 4-х лет.
Актуальность
В мире отмечается значительный рост детей с генетической патологией. Генетические заболевания это болезни, которые возникают в результате различных хромосомных мутаций или наличия дефекта в генах.
Синдром Крузона — это редкое генетическое заболевание, основным проявлением которого является краниосиностоз, т.е преждевременное сращение костей черепа, ведущее к деформации мозгового и лицевого отдела черепной коробки. Впервые это состояние было описано в 1912 году французским педиатром О.Крузоном. Тип наследования — аутосомно-доминантный. Одинаково поражает как мальчиков, так и девочек.
Симптомами этого состояния являются изменение формы головы, крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха.
Выделяют два варианта синдрома Крузона: классический и с черным акантозом.
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме — он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов (более 35 мутаций), что снижает его стабильность. В основном поражает элементы скелета. Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом и к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона — черепного синостоза.
Причины синдрома Крузона с черным акантозом несколько иные — он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями — Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта.
Синдром Крузона с черным акантозом характеризуется аналогичными изменениями со стороны лица и черепа. При этом некоторые исследователи отмечают, что данная форма заболевания протекает в целом тяжелее и характеризуется повышенной частотой осложнений. Так, атрезия хоан, в случае синдрома Крузона с черным акантозом регистрируется почти у половины больных. Кроме того, у пациентов наблюдаются сильно выраженные кожные нарушения — гиперкератоз (разрастание бородавок, гипертрофия кожи), усиленная пигментация в области коленных и локтевых сгибов, шеи, живота, носогубных складок, зоны вокруг глаз; невусы (родинки), часто развиваются гипертрофические слабо пигментированные рубцы и шрамы.
Клинические проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» — в зависимости от того, по какому шву произошло срастание. При пальпации очень хорошо чувствуются уплощенные края швов. Около места соединения стрелковидного и венечного швов черепа зачастую наблюдаются экзостозы. Такая деформация приводит к поражению органов зрения — сначала возникает расходящееся косоглазие, затем экзофтальм вплоть до выпадения глазных яблок из орбиты. Экзофтальм всегда является вторичным. Его развитие обусловлено уменьшением глубины орбит. Поражение зрительного нерва наблюдается у 80% пациентов. В некоторых случаях может наблюдаться эктопия зрачка, колобома радужной оболочки, эктопия хрусталика, мегалокорнеа, спонтанный вывих глазного яблока.
Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости — ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Изменений со стороны вестибулярной системы не обнаруживается.
Наблюдаются изменения и со стороны нервной системы это нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Диагностика синдрома Крузона возможна на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методы, компьютерная томография, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития.
На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов, маленькие параназальные синусы, расширение ямки гипофиза, базилярный кифоз, уплощение глазниц и пальцевые вдавления.
Томографическое исследование четко указывает на деформацию внутреннего слухового прохода. Томография височной кости обнаруживает отсутствие барабанной полости, атрезию или стеноз наружного слухового прохода, сужение и искривление воздухоносных пространств сосцевидного отростка и среднего уха, деформацию стремени, анкилоз молоточка с внешней стенкой верхнего отдела барабанной полости, недоразвитие периостальной части лабиринта.
Молекулярно-генетическая диагностика гена FGFR2 выявляет место мутации и поиск мутации Ala391Glu в гене FGFR3 при наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках).
Мы наблюдали классический вариант синдрома Крузона. К нам в неврологическое отделение ОДММЦ г.Андижана поступила девочка 2013 года рождения с жалобами матери на необычный вид ребенка: пучеглазие, «некрасивую» форму головы, задержку развития, судороги.
Из анамнеза: ребенок от первой беременности, первых родов, родилась в срок с весом 4200г с оценкой по шкале Апгар 4 балла. Беременность протекала на фоне резко выраженного токсикоза в первой половине беременности, угрозы на втором месяце беременности. Мама перенесла грипп в первом и третьем триместрах беременности. Лечение не получала. Брак не родственный. Родители молодые, внешне здоровые. Родственные браки отмечены по линии отца ребенка. Наследственные заболевания отрицают. Родословную составить не удалось, родители в разводе. Состоит на учете у невропатолога с 3-х месячного возраста по поводу гидроцефального синдрома. В 1,5 года выставлен диагноз: Синдром Крузона. Из профилактических прививок получила ВГВ, БЦЖ, ОПВ (в род.доме). Девочке 4 года. Весит 12кг. Рост 82см. Окружность головы 44см. Отмечена задержка физического, нервно-психического и психомоторного развития ребенка. Начала сидеть в течение последних трех месяцев. Ходит с поддержкой. Речь отсутствует, но начала издавать звуки. Не понимает обращенную к ней речь. Пытается следить за движущимися предметами. Периодически нистагм и расходящееся косоглазие. Выраженный экзофтальм(рис.1). Веки не смыкаются. Не моргает. Со слов матери, когда «глаза устают» и во время сна девочка принимает колено-локтевое положение, уткнувшись лицом в подушку. На осмотр и звуки реагирует вяло. Не дифференцирует где свои, где чужие, но мать чувствует. Зрачки D=S, округлой формы. Движения глазных яблок безболезненное. Лицо симметричное. Язык по средней линии. Глотание не нарушено. Парезов и параличей конечностей нет. Менингеальных знаков и патологических рефлексов нет. Чувствительность не нарушена. Координационные пробы провести невозможно.

Рис.1 Выраженный экзофтальм у больной М., 2013года рождения с синдромом Крузона
Кожные покровы чистые от сыпи, нет участков гиперкератоза, отсутствуют невусы, шрамы. Периферические лимфатические узлы не увеличены.
Костно-суставная система. Череп неправильной формы(рис.2). Затылок скошен, вытянут кверху. Выраженный экзофтальм, Уши низко посажены/ Нос маленький. Наблюдается нарушение носового дыхания. Сопит. Дышит ртом. В легких везикулярное дыхание. Тоны сердца ритмичные, слегка приглушены. Язык чистый. Готическое небо. Живот мягкий, пальпаторно безболезненный. Печень и селезенка в пределах возрастной нормы.
Физиологические отправления не нарушены.
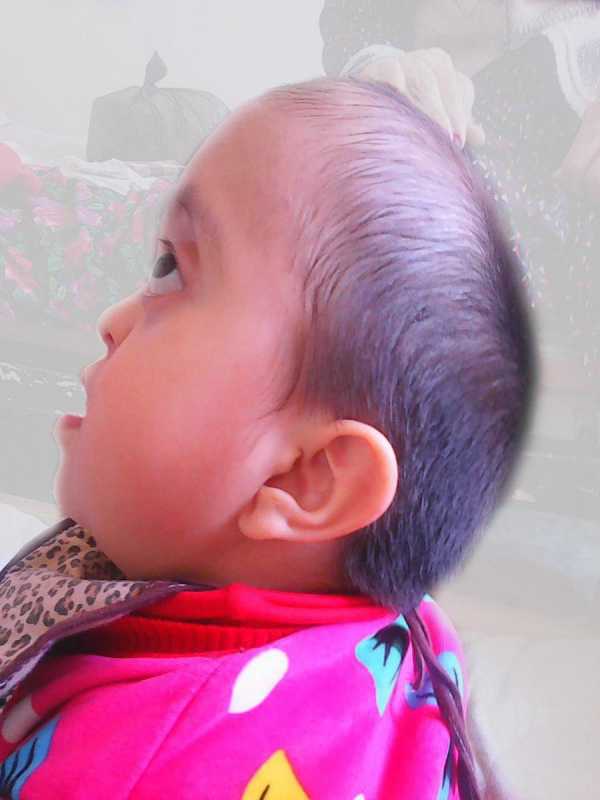 Рисунок 2. Внешние признаки синдрома Крузона у больной М., 2013 года рождения
Рисунок 2. Внешние признаки синдрома Крузона у больной М., 2013 года рожденияДевочка была обследована.
Общий анализ крови: анемия гипохромная средней степени тяжести
Общий анализ мочи: без патологических изменений
ЭКГ: Синусовый ритм правильный. Нормальное положение ЭОС.
На рентгенографии черепа регистрируется неправильная форма черепа, маленькие параназальные синусы, расширение ямки гипофиза, уплощение глазниц, пальцевые вдавления.
Компьютерная томография черепа от 20.04.2018г. Объёмных образований не выявлено. Деформация лицевого скелета. Уплощение глазниц. Экзофтальм. Орбитальный гипертелоризм. Признаки внутричерепной гипертензии.
Осмотрена следующими специалистами
- окулистом: Глазное дно: застойные диски зрительного нерва (за счет повышения внутричерепного давления), 2-х сторонний экзофтальм, расходящиеся косоглазие, нистагм, астигматизм.
- отоларингологом: искривление носовой перегородки слева.
- генетиком: черепно-лицевой дизостоз.
Заключительный диагноз: Синдром Крузона с гипертензионным синдромом. Грубая задержка нервно-психического, психомоторного и физического развития.
Заключение
Уровень медицины нашего времени позволяет своевременно диагностировать генетическую патологию. Для этого необходимо усилить санитарно-просветительную работу среди населения о последствиях родственных браков, путем бесед, показа видеороликов, а также обязательный 100% скрининг перед заключением брака, и беременным женщинам для предупреждения и раннего выявления врожденной и наследственной патологии.
Синдром Крузона
Редкая генетическая патология, при которой наблюдаются деформации лица и черепа, краниосиностоз, а также развиваются сопутствующие нарушения. Заболевание может проявляться брахицефалией, скафоцефалией, тригоноцефалией, гипоплазией средней части лица, крючковидным носом, зрительными и слуховыми нарушениями. Диагноз устанавливают на основании анамнестических данных, клинических симптомов, физикального осмотра и дополнительных обследований. В рамках диагностики могут выполнять автоматическое секвенирование гена FGFR2 и FGFR3, компьютерную томографию, рентгенографию, офтальмоскопию. Специфическая терапия не разработана, потому проводится паллиативное лечение. Деформации лица и черепа исправляют с помощью оперативных вмешательств. Если у пациента выявляется атрезия хоан, чтобы облегчить дыхание, их расширяют хирургическим путем. Прогноз сложный. Большинство врачей склоняются к неблагоприятному исходу. Несмотря на симптоматическую и паллиативную терапию, со временем клиническая картина дополняется нарастающим расходящимся косоглазием, глухотой, выраженной гипоплазией части лица. Возможно развитие олигофрении. Прогноз выживаемости благоприятный. Если больной получает необходимое лечение, он может дожить до старости.
Причины синдрома Крузона
Болезнь развивается на фоне генетических мутаций. Дефектным геном является FGFR2, который располагается в 10-й хромосоме. Он отвечает за кодирование аминокислотной последовательности рецепторов к росту фибробластов. Кроме того, различные мутации данного гена могут привести к формированию акроцефалосиндактилии 1 типа, синдромам Пфайффера, Сетре-Чотзена и многих других заболеваний. Из-за миссенс-мутаций изменяется структура кодируемого протеина. В результате нарушается взаимодействие между клетками соединительных тканей черепа. Фибробласты накапливаются в зоне межкостных швов, а затем костенеют, формируя черепной синостоз. Синдром Крузона с черным акантозом образуется при мутациях другого гена – FGFR3, который локализуется в 4 хромосоме.
Симптомы синдрома Крузона
Признаки недуга заметны сразу после родоразрешения, правда наиболее выраженные симптомы отмечаются в возрасте 4-х лет. Заболевание проявляется краниосиностозом, развивающемся в зоне венечного либо стреловидного шва, который прочно соединяет кости и останавливает рост черепа. Уже в первые дни жизни патология характеризуется гипертелоризмом, крючковидным носом, незначительным экзофтальмом, прогнатием нижней челюсти, низким расположением слухового прохода. У некоторых пациентов болезнь сопровождается синдактилией пальцев. Возможно развитие гидроцефалии и атрезии хоан. Болезнь неминуемо прогрессирует. К симптомам недуга также относят брахицефалию, экзостозы костей черепа, косоглазие, нарушения слуха, умственную отсталость, судороги. При форме синдрома с черным акантозом также наблюдаются гиперкератоз, сильная пигментация, большое количество невусов, гипертрофические рубцы.
Диагностика синдрома Крузона
В большинстве случаев патология выявляется во время проведения пренатальной диагностики. Чтобы становить диагноз после рождения малыша, врач собирает анамнестическую информацию, анализирует клинические признаки, проводит физикальный осмотр и направляет ребенка на дополнительные обследования. В рамках диагностики могут выполнять автоматическое секвенирование гена FGFR2 и FGFR3, компьютерную томографию, рентгенографию, офтальмоскопию. Пациенту могут потребоваться консультации специалистов педиатрического, офтальмологического, оториноларингического, психиатрического, хирургического и генетического профилей.
Лечение синдрома Крузона
Специфическая терапия не разработана, потому проводится паллиативное лечение. Чтобы ремоделировать форму черепа и устранить синостозы, врачи назначают оперативные вмешательства. В большинстве случаев их проводят в несколько этапов по мере роста ребенка. Операции также необходимы для снижения давления внутри черепа, профилактики развития умственной отсталости и неврологических расстройств. Степень экзофтальма снижают с помощью искусственного блефарофимоза. Атрезия хоан является показанием к их хирургическому расширению. Если развиваются кожные изменения, назначают ретиноиды и кортикостероиды в форме мази или крема.
Профилактика синдрома Крузона
Специальных превентивных мер не существует. Парам, планирующим зачатие, рекомендована консультация генетика. В период гестации проводится пренатальная диагностика с помощью молекулярно-генетических исследований.
Синдром Крузона - ДНК-диагностика - Центр Молекулярной Генетики
 Синдром Крузона (craniofacial dysostosis type I) – аутосомно-доминантное заболевание, характеризующееся черепным синостозом, гипертелоризмом, экзофтальмом и наружным косоглазием. Нос больных напоминает клюв попугая. Также у больных отмечается короткая верхняя губа, гипопластичная верхняя челюсть и выступающая нижняя челюсть. Семейные случаи заболевания впервые описаны Крузоном в 1912 году.
Синдром Крузона (craniofacial dysostosis type I) – аутосомно-доминантное заболевание, характеризующееся черепным синостозом, гипертелоризмом, экзофтальмом и наружным косоглазием. Нос больных напоминает клюв попугая. Также у больных отмечается короткая верхняя губа, гипопластичная верхняя челюсть и выступающая нижняя челюсть. Семейные случаи заболевания впервые описаны Крузоном в 1912 году.
Синдром Крузона составляет примерно 4.8% случаев с черепным синостозом, выявляемых при рождении. Частота заболевания оценивается как 16.5: 1 000 000 новорожденных. Отмечается эффект возраста отца при образовании мутации de novo.
К синдрому Крузона приводят мутации в гене рецептора фактора роста фибробластов-2FGFR2 (fibroblast growth factor receptor 2; OMIM 176943). Ген FGFR2 локализован на хромосоме 10q26 и состоит из 20 экзонов. Мутации, вызывающие развитие этого синдрома, в основном располагаются в экзонах 7 и 9 гена. Всего в гене FGFR2 выявлено 35 различных мутаций, приводящих к заболеванию. При исследовании происхождения мутаций de novo с помощью внутригенных полиморфных маркеров во всех информативных случаях показано их отцовское происхождение.
К Crouzon syndrome с acanthosis nigricans (синдрому Крузона с черным акантозом) приводит мутация в гене FGFR3 (MIM 134934) p.391 Ala>Glu.
В Центре Молекулярной Генетики проводится поиск мутаций в экзонах 7 и 9 гена FGFR2 методом прямого автоматического секвенирования.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Крузона синдром
Синдром Крузона Симптомы, причины, лечение / нейропсихология | Thpanorama
Синдром Крузона это черепно-лицевой порок развития, вызванный ненормальным закрытием или развитием черепных швов и, как следствие, вызывает различные аномалии на лице и черепе (Shneider et al., 2011).
Это патология врожденного происхождения, связанная с наличием частичной или полной мутации гена FGFR2, связанной с фактором роста фибробластов (FGFR) (Seattle Children's Hospital, 2016).
Клинически, синдром Крузона характеризуется наличием выпуклости или выпуклости лобной части черепа, укорочением общего объема головы, гипоплазией верхнечелюстной кости или нормальным развитием глазных впадин, среди других аспектов (Бостонская детская больница, 2016).
Что касается диагноза, клинические признаки обычно не четко видны во время рождения. Как правило, физические характеристики проявляются примерно в два года. Таким образом, диагноз подтверждается на основании подробного физического обследования и генетического исследования (Orphanet, 2013).
Хотя нет никакого лечения от синдрома Крузона, существует широкий спектр терапевтических подходов, которые могут значительно улучшить медицинские осложнения, связанные с этой патологией..
Во всех случаях выбор лечения основывается на работе многопрофильной команды: стоматологии, нейрохирургии, офтальмологии, травматологии, физиотерапии, логопедии, нейропсихологии и т. Д. (Ассоциация челюстно-лицевых пороков развития и аномалий, 2012).
Характеристика синдрома Крузона
В частности, эта патология была впервые описана в 1912 году хирургом французского происхождения Октави Крузоном (Beltrán, Rosas and Jorges, X).
Уже в первых клинических случаях, описанных в медицинской и экспериментальной литературе, удалось обнаружить явную связь черепно-лицевых признаков с аномальным образованием черепных швов (Beltrán, Rosas and Jorges, X).
Самые современные утверждения об этой патологии определяют ее как генетическое расстройство, возникающее в результате краниосиностоза или раннего закрытия костей, из которых состоит череп (Genetics Home Reference, 2016).
Конфигурация черепа во время младенческой или развивающейся стадии имеет овальную структуру, будучи более широкой в задней области. Таким образом, костные части (затылочная, височная, теменная и лобная) обычно образуются на пятом месяце беременности и представлены вместе соединительной или фиброзной тканью, черепными швами (Villareal Reyna, 2016).
Следовательно, черепные швы позволяют наращивать объем головы и мозга благодаря своей гибкости. Кроме того, его закрытие начинает постепенно развиваться между 9 и 24 месяцами (Villareal Reyna, 2016).
Когда происходит изменение этого процесса, такое как краниостеноз, происходит раннее закрытие этих волокнистых структур (Villareal Reyna, 2016).
Таким образом, это событие препятствует нормальному формированию структуры черепа, лица и мозга. Как следствие, у больного человека развивается множество пороков развития, которые влияют на глаза, положение челюсти, форму носа, зубы или образование губ и неба (Genetics Home Reference, 2016).
Несмотря на то, что у большинства людей с синдромом Крузона наблюдается нормальная или ожидаемая потребность в возрастной группе, привычное развитие мозга может быть замедлено и, как следствие, могут возникнуть различные трудности в обучении, которые вместе с аномалиями Зубные и верхнечелюстные зубы значительно замедляют овладение языком (Genetics Home Reference, 2016).
В дополнение к наиболее часто используемому термину «синдром Крузона», эта патология может также иметь ссылки на другие типы названий: краниостеноз типа Крузона, краниофациальный дизостоз или краниофациальный дизостоз Крузона (Национальная организация редких заболеваний, 2007)..
статистика
Частота синдрома Крузона была оценена примерно в 16 случаев на миллион новорожденных во всем мире (Genetics Home Reference, 2016).
В частности, Больница Сиэтла Чиндре (2016) утверждает, что синдром Крузона является патологией, которая может возникать у 1,6% людей на 100 000 человек..
Кроме того, это одна из наиболее частых патологий, обусловленных краниосинтозом. Приблизительно 4,5% людей, которые страдали от краниосинтеза, страдают от синдрома Крузона (Seattle Children's Hospital, 2016).
С другой стороны, что касается распространенности по половому признаку, статистические данные, которые указывают на значительное увеличение числа случаев в любом из них, не были найдены. Кроме того, возникновение синдрома Крузона не было связано с конкретными географическими регионами или конкретными этническими группами.
Симптомы и симптомы
Клинические характеристики и типичные медицинские осложнения синдрома Крузона могут значительно различаться у пострадавших лиц. Однако кардинальным открытием во всем является наличие краниосиностоза.
краниосиностозы
Авторы, такие как Sanahuja et al. (2012), определяют краниосиностоз как патологическое событие, которое приводит к раннему слиянию одного или нескольких черепных швов..
Таким образом, развитие черепа деформируется, растя в направлении, параллельном пораженным областям, то есть рост замедляется в сросшихся швах и прогрессивно продолжается в открытых (Sanahuja et al., 2012).
При синдроме Крузона закрытие черепных костных пластинок происходит в возрасте 2–3 лет до рождения, однако в других случаях это может быть очевидным во время рождения (Сиэтлская детская больница, 2016 г.).
Кроме того, степень вовлеченности может быть переменной, в зависимости от областей или швов, затронутых слиянием.
В наиболее серьезных случаях можно наблюдать слияние швов костных частей, которые образуют лоб и верхние боковые стороны черепа, то есть коронального и сагиттального швов, с одной стороны, и теменных швов с другой. Кроме того, в других случаях также можно обнаружить шов задних отделов задней кости (Национальная организация по редким заболеваниям, 2007).
Таким образом, краниосиностоз является этиологическим событием, которое вызывает остальную часть симптомов и медицинских осложнений, типичных для синдрома Крузона (Национальная организация по редким заболеваниям, 2007).
Черепных пороков
Слияние швов черепа может привести к
продолжительность жизни, лечение и прогноз
- Главная
- Анкилозирующий спондилоартрит
- Pages
- новости здоровья
- Воспитание
- симптом
- Здоровье слайд-шоу
- карты тела человека
- оценка состояния здоровья
- Здоровье
- Пробиотики и пищеварительной здоровья
- Простуда и грипп
- Endotough
- опухоль головного мозга
- Cic
- Halobetasol
- Болезнь сердца
- Бессонница
- Прыщи
- Сухость глаз
- Понимание и.т.п.
- Понимание Гемофилия
- Колоректальный рак
- кожа
- гипотиреоз
- Ваше расширенное путешествие рак молочной железы
- Гиперактивный мочевой пузырь
- Простуда
- Каждый день фитнес
- Рабочее здоровье
- Рак полости рта
- аутизм
- Управление идиопатический легочный фиброз
Синдром Крузона: Симптомы, лечение и перспективы
При синдроме Крузона некоторые кости черепа сливаются слишком рано. Этот процесс называется краниосиностоз. Существуют и другие последствия этого состояния и способы их преодоления.
В этой статье рассматриваются симптомы, методы лечения и перспективы развития синдрома Крузона.
Какие симптомы?
Краниосиностоз обычно не ассоциируется с генетическим синдромом. В редких случаях, когда это вызвано генетикой, это может быть синдром Крузона.
Она затрагивает примерно 16 из каждых миллионов новорожденных детей.
Крузоновый синдром вызывает преждевременное слияние некоторых костей черепа и влияет на форму головы и лица человека.
Признаки синдрома Крузона включают в себя:
- аномальная форма лица
- неглубокая посередине лица, что может привести к затруднению дыхания.
- высокий лоб
- широко расставленные, выпуклые глаза.
- неглубокая глазная впадина, которая может привести к проблемам со зрением.
- глаза, указывающие в разные стороны (косоглазие).
- маленький клювоподобный нос.
- недостаточное развитие верхней челюсти, что может привести к проблемам с приемом пищи.
- стоматологические проблемы
- недоступные уши
- потеря слуха с возможной узкими ушными каналами
Менее распространенным признаком является отверстие в крыше рта (расщелина нёба) или губы (расщелина губы).
Для одних детей симптомы являются тяжелыми, для других — более легкими. Состояние влияет на каждого человека по-своему.
Осложнения
Около 30 процентов детей развивается гидроцефалия, которая влияет на приток жидкости в мозг и позвоночный канал.
Это может привести к повышению давления в черепе (внутричерепное давление), что может повлиять на развитие мозга и вызвать трудности в обучении.
Еще одним осложнением является затрудненное дыхание, которое может представлять опасность для жизни, если его не лечить.
Любому человеку с синдромом Крузона, подверженному риску возникновения внутричерепного давления и затрудненного дыхания, может потребоваться операция.
Причины
В этом разделе рассматриваются гены и их влияние на кости головы:
Гены
Синдром Крузона — это генетический. Это не вызвано тем, что происходит во время беременности.
Это заболевание вызвано мутациями в генах, называемых FGFR2. По мере роста эмбриона этот ген контролируется, когда незрелые клетки становятся костными клетками.
При синдроме Крузона мутация гена FGFR2 влияет на белок FGFRR, вызывая его аномальное поведение в некоторых растущих костях черепа.
Это приводит к преждевременному сращиванию волокнистых суставов (швов) между этими костями.
У кого-то с синдромом Крозона 50% шансов передать это заболевание своему ребенку.
Синдром Крузона не всегда наследуется. Некоторые дети рождаются с таким заболеванием и первыми в своих семьях страдают от этого синдрома. Когда это происходит, это называется de novo мутацией.
диагностирование
Симптомы обычно появляются в первый год жизни ребенка. Иногда симптомы прогрессируют до достижения ребенком возраста 2-3 лет. В других случаях это состояние проявляется при рождении.
Если врач подозревает наличие синдрома Крозона, он проведет медицинский осмотр и задаст вопросы о семейной истории болезни.
Врач может провести дополнительные обследования, в том числе
- рентгеновские лучи
- магнитно-резонансная томография (МРТ) сканирует
- компьютерная томография (КТ).
- генетическое исследование
Варианты лечения
Синдром Крузона по-разному влияет на каждого ребенка.
Лечение синдрома Крузона может включать хирургическое вмешательство. Это необходимо для улучшения симптомов, предотвращения осложнений и содействия физическому и психическому развитию.
Если расплавленный шов вызывает внутричерепное давление, это может привести к черепно-мозговой травме.
Это лечение черепно-лицевой хирургией или операцией в открытом хранилище.
Целью операции является обеспечение достаточного пространства в голове для роста мозга. Это также может улучшить внешний вид лица и головы ребенка.
Если у человека возникают проблемы с дыханием из-за нарушений в работе лица, ему может потребоваться трахеостомия, которая является хирургической процедурой, чтобы сделать отверстие в дыхательной трубке, чтобы помочь ему дышать.
Наряду с хирургическим лечением, люди с синдромом Крозона и их семьи могут получить генетическую консультацию и поддержку. Эти методы лечения часто помогают им справляться с психологическими последствиями жизни с генетическим заболеванием.
Предотвращение
Исследования по предотвращению генетических мутаций, вызывающих синдром Крузона, продолжаются.
Проведенное в 2006 году исследование с использованием животных клеток показало, что потенциальное лечение является перспективным с точки зрения профилактики. Эта процедура направлена на предотвращение сшивания швов в черепе, сливающихся в утробе матери.
Это лечение еще не полностью разработано и не протестировано у животных или людей.
Чаепитие
После хирургического лечения черепно-мозговых нарушений риск осложнений снижается.
После лечения большинство детей с синдромом Крузона продолжают вести здоровый образ жизни.
При лечении это заболевание не влияет на ожидаемую продолжительность жизни.
Синдром Крузона: симптомы, причины :: SYL.ru
Многие патологические процессы, особенно передающиеся на генетическом уровне, определяются при визуальном осмотре. К таковым медицинские работники относят синдром Крузона или черепно-лицевой дизостоз. Это достаточно редкое наследственное заболевание, характеризующееся преждевременным сращиванием швов черепа. Говоря доступным для обывателя языком, наше вместилище головного мозга состоит из многочисленных косточек, которые по мере взросления соединяются между собой как пазлы, образуя швы.
В некоторых случаях наблюдается задержка зарастания в определенных местах, что приводит к деформациям в других частях костей черепа (меняется форма). Такое состояние называется краниосиностозом. Пораженный коронарный шов (от одного уха до второго) приводит к аномалиям костей глазниц (они становятся выпуклыми), меняется высота лба, челюстная мускулатура. Какова точная частота встречаемости этого недуга, медикам пока неизвестно.
На сегодняшний день на территории нашей страны ежегодно появляется один новорожденный из 10 000 детей с врожденными деформациями лицевых и мозговых отделов. Целый ряд косметических уродств, которые характерны для дизостоза, зачастую сочетается с интеллектуальной задержкой либо умственной отсталостью. Бесспорно, это негативно отражается на качестве жизни и здоровье человека. Будем выяснять, каковы факторы, влияющие на развитие болезни, и возможно ли полностью излечиться.
Синдром Крузона: причины возникновения
Многочисленные клинические и лабораторные исследования показали, что основной путь передачи генетического нарушения — аутосомно-доминантный. Это значит, что если у одного из родителей (прародителей) имеется мутирующий ген, то вероятность появления на свет ребенка с подобными расстройствами 50%. Дети, не унаследовавшие дефектные хромосомы, не будут его носителями. Для выявления аномального гена в крови молодой паре необходимо пройти обследование перед зачатием ребенка.
Синдром Крузона: симптомы и проявления
Наблюдаются клинические деформации со стороны костной ткани. Форма коробки зависит от того, какие черепные швы подвержены патологическому процессу. У некоторых пациентов голова имеет вид треугольника, у других — слишком удлиненная и узкая. Кроме того, черепная коробка может быть увеличена диаметрально относительно продольной части. Синдром Крузона провоцирует значительные изменения головы.
При ручном обследовании обнаруживаются костные образования (уплотнения) в области родничка, затылочной части и ушных раковин. Четко прослеживается недоразвитость верхней губы и челюсти. Наблюдается клювовидный заостренный нос, редкий зубной ряд (неполное смыкание), торчащий язык, высокое нёбо, расщелина на язычке. У ребенка отмечается задержка речи, интеллекта. Нарушения сохраняются на протяжении всей жизни, затрудняя межличностное общение в социуме.
Какие изменения наблюдаются со стороны зрения и слуха?
Экзофтальм или смещение глазных яблок является неотъемлемым признаком при черепно-лицевом дизостозе. Это обусловлено значительным уменьшением радиуса глубины орбит. Визуально видно, что глаза сильно выпучены, раскосые, постоянно вращаются (спонтанные движения). Со стороны выглядит как нервный тик.
Нередко синдром Крузона вызывает эктопию зрачков, хрусталика и радужной оболочки — отклонение от центрального месторасположения. Значительно нарушается зрение. По медицинским данным, около 30% больных страдают глухотой. При осмотре выявляется зарастание слухового прохода.
Лабораторные исследования
В первую очередь доктор осматривает пациента, тщательно изучает анамнез патологии, выясняет наследственный фактор. Не составляет сложности диагностировать синдром Крузона. Фото заболевания ярко иллюстрирует лицо больного с подобными расстройствами. Обязательно проводится рентгенография черепной коробки.
На снимке будут видны срощенные швы и форма головы. Также заметны повреждения в околоносовых пазухах, уплотнения в глазных яблоках, искривление головного мозга. Рентген поможет распознать имеющиеся деформации в слуховом проходе.
Назначается компьютерная и магнитно-резонансная томография. С помощью этих методов обнаруживается стеноз слухового прохода, отсутствие барабанной перепонки и прочие аномалии. Врач может направить пациента к узкопрофильным специалистам: генетику, невропатологу, нейрохирургу, офтальмологу.
Какова терапия
К сожалению, синдром Крузона не излечивается. Единственное, что могут предложить врачи - косметическую либо функциональную коррекцию синостозированных швов. При помощи пластической хирургии возможно восстановить строение лицевой части черепа, структуру мягких тканей и костей, ликвидировать эффект «выпученных глаз» и выровнять носовую перегородку.
Современная медицина дает шанс на полноценное существование, избавление от физических уродств, препятствующих нормальному общению. Хирургическую терапию следует выполнять на начальном этапе развития — в раннем детском возрасте, пока кости черепа не успели срастись и деформироваться. После оперативного вмешательства больному требуется какое-то время носить фиксирующий шлем, который предотвратит травмы.
Прогноз
Даже опытный врач не сможет дать никаких гарантий в отношении этого заболевания даже после проведения операции. Очень часто вызывает полную потерю зрения синдром Крузона. Что это за «страшный зверь», который не поддается лечению, специалисты пока пытаются понять и изучить. Но до сих пор этот недуг захватывает человечество и наносит непоправимый ущерб. С возрастом болезнь прогрессирует. Значительные деформации наблюдаются в области лица.
Однако человек может длительное время сохранять трудовую и социальную адаптацию, несмотря на косметические недостатки. Самое обидное, что дизостоз невозможно искоренить, не существует мер по его профилактике, не создано медикаментозных средств. Будем искренне надеяться на прогрессивные медицинские технологии, которые когда-нибудь помогут избавить людей от генетических расстройств.
синдром Крузона - Crouzon syndrome
Крузона синдром является аутосомно - доминантное генетическое заболевание известно как жаберной дуги синдрома. В частности, этот синдром влияет на первой жаберной (или глоточной) арка, которая является предшественником верхней челюсти и нижней челюсти . Поскольку жаберная арка являются важными особенностями развития в растущем эмбрионе , нарушение их развития создает прочное и снотворное действие.
Этот синдром назван в честь Octave Крузона , с французским врачом , который впервые описал это заболевание. Он отметил , что пострадавшие пациенты были мать и ее дочь, подразумевая генетическую основу. Первый называется «краниофациальный дизостоз», расстройство характеризуется рядом клинических признаков. Этот синдром вызывается мутацией в рецепторе фактора роста фибробластов 2 ( FGFR2 ), расположенном на хромосоме 10 .
Ломая название « черепно - лицевой » относишься к черепу и лицу , и « дизостозам » относится к уродству кости. В настоящее время известно как синдром Крузона, характеристики могут быть описаны зачаточными значениями его прежнего названия. Что происходит в том , что череп и кости лица неопытного младенца, в то время как в разработке, взрывателя рано или не могут расширяться. Таким образом, нормальный рост костей не может произойти. Слияние различных швов приводит к аномальным формам роста черепа.
Признаки и симптомы
Ребенок с синдромом Крузона показывая характерные черты лица.Отличительная черта синдрома Крузона является краниосиностозом , что приводит к аномальной форме головы. Это присутствует в комбинации: акроцефалии , лобной начальствование , тригоноцефалии (слияния метопического шва ), брахицефалия (слияние венечного шва), долихоцефалия (слияние сагиттального шва ), плагиоцефалия (односторонний преждевременное закрытие ламбдовидных и корональных швы ) , oxycephaly (слияние корональных и lambdoidal швов ), а также комплекс краниосиностоз (преждевременное закрытие некоторых или всех швов).
Экзофтальм (выпуклые глаза из - за мелкую глазницу после раннего слияния окружающей кости), гипертелоризм (больше , чем нормальное расстояние между глазами) и psittichorhina (клювом , как нос), также очень общие чертами. Другие лица характеристики, которые присутствуют во многих случаях включает в себя внешнее косоглазие и гипоплазии тахШага (недостаточный рост зоны лица), что приводит к относительному нижнечелюстному прогнатизму (выступающий подбородок) и дает эффект у пациента , имеющий вогнутую поверхность.
Большинство симптомов являются вторичными по отношению к аномальной структуре черепа. Около 30% людей с синдромом Крузона развивается гидроцефалия . Нейросенсорная тугоухость присутствует в некоторых случаях. Аномалии в порядке , в котором глаз вписываться в глазницах могут вызвать проблемы со зрением. Некоторые люди с условием имеют ограниченные дыхательные пути и могут возникнуть серьезные проблемы с дыханием.
Синдром Крузона также связан с патентом артериального протока (PDA) и коарктацией аорты .
Общие черты являются узким / высоким арочным небом, задним двусторонний прикус, hypodontia (отсутствуют некоторые зубы), и скученность зубов . В связи с верхнечелюстной гипоплазии, люди с синдромом Крузона как правило , имеют значительную постоянную underbite и впоследствии не может жевать с помощью резцов.
По причинам, которые не совсем ясны, большинство пациентов Крузона также заметно короче плечевой кости и бедренной кости по отношению к остальной части их тела , чем члены населения в целом. Небольшой процент пациентов Крузона также имеет то , что называется «Тип II» синдром Крузона, отличается частичной синдактилией .
причины
Современные исследования показывают , фактор роста фибробластов рецепторов (FGFR) Fgfr2 и FGFR3 , как ведущих факторов вызывая аутосомно - доминантный синдром Крузона. Эти два трансмембранные белками являются двумя из четырех факторов роста фибробластов рецепторов , участвующих в остеобластах дифференциации во время эмбрионального развития ; мутации среди этих рецепторов участвуют в нескольких генетических нарушений. Есть 40 известных мутаций, большинство из которых вызваны мутацией миссенса. FGFR2 является наиболее часто мутированным геном, миссенс на цистеина 342 в экзоне 9, которая создает выигрыш-о-функции. FGFR2lllc изоформы , созданная с помощью альтернативного сплайсинга экзона 3 гена FGFR2, использует экзон 9 и используется в мезенхимальных стволовых клеток для управления оссификации . Тем не менее, мутация конститутивно активирует белка трансмембранных через дисульфидную связь , образованную неправильно из - за потери цистеина 342. FGFR3 выражена в лобных костях во время эмбрионального развития, направляя развитие черепной кости. Мутация точки вызывает конститутивную активацию тирозина в петле активации, расположенной в цитозоле области белка, что приводит к ускоренной дифференциации остеобластов лобных, что приводит к преждевременному слиянию лобных костей черепа.
диагностика
Диагностика синдрома Крузона обычно может происходить при рождении по оценке физического облика ребенка. Дальнейший анализ, в том числе рентгенограммы, магнитно - резонансная томография (МРТ) сканирование, генетическое тестирование, рентгеновские лучи и КТ могут быть использованы для подтверждения диагноза синдрома Крузона.
лечение
 Аномальное слияние костей черепа характерен синдром Cruzon.
Аномальное слияние костей черепа характерен синдром Cruzon. Каждый ребенок индивидуален и полностью зависит от того, сливают швы и как это влияет на ребенок о том, как это лечится. Некоторые дети имеют серьезные проблемы с дыханием из-за мелкой середины лица и может потребовать трахеостомии.
Хирургия обычно используется для предотвращения закрытия швов черепа от повреждения развития мозга. Без хирургического вмешательства, слепота и интеллектуальная инвалидность являются типичными результатами. Черепно - лицевая хирургия является дисциплиной как пластической хирургии и челюстно-лицевой хирургии (OMFS). Для перемещения орбиты вперед, краниофациальные хирурги подвергать черепа и орбиты и изменения формы кости. Для лечения дефицита зоны лица, хирурги могут двигаться нижние орбиты и средней зоны кости вперед ..
Крузона пациенты , как правило, имеют несколько швов , связанных, в частности , большинство двусторонних корональные craniosynostoses , и либо операции на открытом хранилище или полоса удаление фрагментов костей черепа (если ребенок в возрасте до 6 месяцев) может быть выполнена. В последнем случае, шлем надет в течение нескольких месяцев после операции.
После обработки для черепно-мозговых нарушений сводов, пациенты Крузона обычно идут на жить нормальную продолжительность жизни.
эпидемиология
Заболеваемость синдрома Крузона в настоящее время оценивается в 1,6 происходит из каждых 100000 людей и является наиболее распространенным синдромом craniostenosis.