Синдром леопарда что это такое
Синдром LEOPARD. Симптомы, диагностика, лечение
Синдром LEOPARD является комплексом дисморфогенетических расстройств с переменной пенетрантностью и экспрессивностью. Горлин впервые представил аббревиатуру LEOPARD в качестве названия этого синдрома в 1969 году. Буквы этой аббревиатуры описывают основные черты этого расстройства, а именно:
- Лентиго (L)
- Электрокардиографические нарушения проводимости (E)
- Глазной гипертелоризм (O)
- Стеноз легочной артерии (P)
- Аномалии половых органов (A)
- Замедление роста (R)
- Глухота (D)
Зейслер и Беккер первыми описали этот синдром в 1936 году, провев обследование 24-летней женщины с прогрессивным лентиго, гипертелоризмом и прогнатизмом. Вскоре были зафиксированны первые семейные случаи у близнецов Розен, а затем еще 8 человек из большой семьи. Монахан был первым, кто составил документальную ассоциацию синдрома с сердечными аномалиями и низкорослостью в 1962 году.
Синдром LEOPARD. Причины
Молекулярные исследования показали, что синдром LEOPARD является аллельным расстройством, которое вызывается различными мутациями в гене PTPN11, кодирующем протеин-тирозин-фосфатазу SHP-2, этот ген расположен в полосе 12q24.1. В 2005 году, Огата и Йошида документально подтвердили, что мутации в гене PTPN11 могут быть идентифицированы приблизительно у 80% больных с синдромом LEOPARD.
Еще один интересный случай синдрома LEOPARD был обнаружен в одной семье, у отца и его взрослого сына. Отец имел лентиго, которое было одинаково распределено по всему телу, в то время как у его сына лентиго отсутствовало на левой части грудной клетки, на спине, на левой и правой руках. Позже было установленно, что сын имел мозаичный кариотип в лимфоцитах (47, XXY / 46XY). При анализе тканей полученных на биопсии пигментированной кожи, в основном прослеживался кариотип 47, XXY в то время как кариотип 46, XY прослеживался в непигментированных областях. Некоторые исследователи пришли к выводу, что такие факторы как мозаичность, типы мутаций в гене PTPN11, а также кол-во и характеристики половых хромосом могут влиять на фенотип синдрома LEOPARD.
Синдром LEOPARD. Фото
Лентиго на лице ребенка с синдромом LEOPARD.
Лентиго на склере у ребенка с синдромом LEOPARD.
Неупорядоченная пигментация на туловище у пациента с синдромом LEOPARD.
Ониходистрофия
Синдром LEOPARD. Симптомы и проявления
Высоко вариабельная экспрессивность синдрома делает его диагностику трудной задачей, особенно в спорадических случаях. Семьдесят процентов случаев являются семейными. На основании клинического анализа большой серии пациентов, собранных в медицинской литературе, в 1976 Ворон вывел минимальные критерии для диагностики. Для синдрома LEOPARD обязательно наличие лентиго и по крайней мере двух из следующих проявлений и знаков:
- Другие кожные нарушения
- Сердечные структурные аномалии
- Аномалии в мочеполовой системе
- Эндокринные нарушения
- Неврологические дефекты
- Дисморфизмы лица
- Дефицит роста
- Скелетные аномалии
Если лентиго отсутствует, то диагноз синдрома LEOPARD можно поставить при наличии минимум трех из вышеуказанных знаков и проявлений. Диагностика синдрома LEOPARD сильно затрудненна у маленьких детей. Диагноз может быть клинически подозреваемым в первые месяцы жизни у пациентов, имеющих минимум три основных проявления: характерные черты лица (100%), гипертрофическая кардиомиопатия (87%) и кожные пятна цвета кофе с молоком (лентиго [75%]) .
Физическое обследование
Лентиго – мелкие, темно-коричневые, полигональные, неправильной формы пятна, размером, как правило, 2-5 мм в диаметре, но иногда и больше, до 1-1,5 см. Эти пятна часто присутствуют на лице, шее и на верхней части части туловища, на ладонях, ступнях и на склерах. Лентиго являются наиболее заметным проявлением синдрома LEOPARD. Оно присутствует у более чем 90% пациентов. Однако, отсутствие этих пятен не исключает наличия этого синдрома. При тщательном обследовании кожи, можно обнаружить другие кожные нарушения, например:
- Подмышечные веснушки
- Локализованные гипопигментации
- Ониходистрофии
- Гиперупругую кожу
Умственная отсталость, обычно легкой степени, наблюдается примерно у 30% пациентов. Около 25% пациентов имеют нейросенсорную потерю слуха. Судороги, нистагм или гипосмии были зарегистрированы у нескольких пациентов. Треть пациентов имеют невысокий рост, который станет очевидным, вскоре после рождения (большинство новорожденных имеют нормальный вес при рождении).
Несмотря на частое развитие пороков сердца, большинство пациентов остаются бессимптомными. Однако, в некоторых случаях, пациенты могут проявлять признаки патологий в сердце. Интересно, что более высокая частота семейной истории внезапной смерти и фибрилляции предсердий сообщалась у пациентов с гипертрофией левого желудочка без мутаций в гене PTPN11.
Около 35% пациентов демонстрируют различные черепно-лицевые аномалии. Глазной гипертелоризм является наиболее встречаемой лицевой аномалией (25%). Другие признаки включают:
- Нижнечелюстной прогнатизм
- Широкий носовой корень
- Дисморфозмы черепа
- Низкий посаженные уши
- Стоматологические аномалии
- Высокое небо
- Кожные складки
- Птоз
- Опухоли роговицы
Аномалии развития мочеполовой системы описаны у 26% больных, преимущественно у мужчин. Аномалии наружных половых органов, такие как крипторхизм или гипоспадия, можно обнаружить на медосмотре. Различные типы скелетных аномалий могут включать деформации грудной клетки, кифосколиоз, аномалии ребер, синдактилии, задержки развития или агенез постоянных зубов, сверхкомплектные зубы. Синдром LEOPARD может быть связан с гипертрофией сплетений, что может привести к невропатической боли.
Синдром LEOPARD. Диагностика
- КТ или МРТ головы
- Рентгенография скелета
- Эхокардиография
- Обследование мочеполовой системы
- УЗИ
- ЭКГ
Синдром LEOPARD. Лечение
Криохирургия и лазерное лечение могут быть полезными в лечении изолированных лентиго. Однако, из-за большого количества пятен лентиго, эти подходы могут быть очень трудоемкими. Для некоторых пациентов, лечение кремом третиноином и гидрохиноном может быть полезным.
Для лиц, больных структурными сердечными аномалиями, терапевтические схемы могут включать блокаторы бета-адренергических рецепторов или блокаторы кальциевых каналов.
Антиаритмические препараты могут потребоваться в случаях развития опасных для жизни желудочковых эктопий.
Хирургическое вмешательство может быть необходимым в случаях тяжелых деформаций и аномалий.
Синдром LEOPARD. Осложнения
Осложнения могут возникнуть из-за связанных аномалий.
Синдром LEOPARD. Прогноз
Прогноз в основном определяется по наличию сердечно-сосудистых осложнений. Большинство пациентов с синдромом LEOPARD могут вести нормальную жизнь. Сердечные патологии могут быть причиной смерти некоторых пациентов.
Consilium Medicum - Consilium Medicum
7 апреля, 2020
Пятый юбилейный Медицинский форум-выставка «Неделя здравоохранения в Республике Башкортостан»Месторасположение:Уфа, ВДНХ ЭКСПО
7 апреля, 2020
XXVI Всероссийский конгресс с международным участием и специализированной экспозицией «Амбулаторно-поликлиническая помощь в эпицентре женского здоровья от менархе до менопаузы»Месторасположение:Москва, ул. Академика Опарина, д.4, ФГБУ «НМИЦ АГП им. В.И. Кулакова» Минздрава России
7 апреля, 2020
IV-й Международный конгресс, посвященный А.Ф. Самойлову «Фундаментальная и клиническая электрофизиология. Актуальные вопросы аритмологии»Месторасположение:Казанская Ривьера, г. Казань, пр. Фатыха Амирхана, 1
8 апреля, 2020
Конференция «Кардиопульмонология 2020»Месторасположение:Центральный Дом Журналиста, Россия, Москва, Никитский бульвар, 8А (м. Арбатская)
9 апреля, 2020
Всероссийская научно-практическая конференция с международным участием "Эпидемиологическая безопасность медицинской деятельности"Месторасположение:«Конгресс-холл Торатау» Республика Башкортостан, Уфа, ул. Заки Валиди, 2)
9 апреля, 2020
IV Общероссийскую конференцию с международным участием «FLORES VITAE. Педиатрия и неонатология»Месторасположение:«AZIMUT Отель Олимпик Москва», Москва, Олимпийский проспект, 18/1.
9 апреля, 2020
Всероссийская конференция с международным участием "Актуальные вопросы трансфузиологии"Месторасположение:г. Санкт-Петербург, Лодейнопольская ул., 5, Конгрессный центр ПетроКонгресс
Леопард синдром: множественный лентигиноз и гипертрофическая кардиомиопатия. Случай из практики
Ukrainian Journal of Perinatology and Pediatrics. 2019. 4(80): 93-98; doi 10.15574/PP.2019.80.93
Мальская А. А.1, Куриляк О. Б.2
1Львовский национальный медицинский университет имени Даниила Галицкого, Украина
2КНП ЛОР «Львовская областная детская клиническая больница «ОХМАТДЕТ», Украина
Для цитирования: Мальская АА, Куриляк ОБ. (2019). Леопард синдром: множественный лентигиноз и гипертрофическая кардиомиопатия. Случай из практики. Украинский журнал Перинатология и Педиатрия. 4(80): 93-98. doi 10.15574/PP.2019.80.93
Статья поступила в редакцию 03.07.2019 г., принята в печать 18.11.2019 г.
Леопард синдром (ЛС) — аутосомно-доминантное заболевание, характеризующееся множественными лентиго и пятнами «кофе с молоком», нарушениями на электрокардиограмме (ЭКГ), глазным гипертелоризмом / обструктивной кардиомиопатией, стенозом легочной артерии, патологией гениталий (у мужчин), задержкой роста и глухотой. Частота данного синдрома неизвестна, в литературе описано около 200 клинических случаев.
Клинический случай. Представлен клинический случай девочки с ЛС, госпитализированной в отделение КНП ЛОР «Львовская областная детская клиническая больница «ОХМАТДЕТ» с жалобами на трудности в обучении, по сравнению со сверстниками, быструю утомляемость, головные боли, одышку и учащенное сердцебиение при физической нагрузке. При осмотре выявлено состояние средней тяжести. Кожа и слизистые оболочки бледно-розовые, гипертелоризм глаз, множественные пигментированные веснушки, распространенные по всему телу, гипергидроз ладоней. Физическое развитие ниже среднего, дисгармоничное по росту. Периферические лимфатические узлы не увеличены. В легких — везикулярное дыхание. Хрипы не выслушиваются. Аускультативно тоны сердца приглушены, ритмичные, частота сердечных сокращений — 90 уд./мин, интенсивный (4/6) систолический шум в 2–4-м межреберье слева и на верхушке. Артериальное давление — 110/60 мм. Во время комплексного обследования, ЭКГ диагностированы признаки систолической перегрузки и гипертрофии левого желудочка, а также признаки ишемии задней и боковой стенки левого желудочка. При Эхо-кардиографическом обследовании диагностирована гипертрофическая кардиомиопатия, асимметрическая, обструктивная форма с внутрисердечным градиентом — 66–106 мм рт. ст. На консультации у ЛОР-врача диагностирована двусторонняя сенсоневральная приглуховатость II степени, более выраженная слева. Консультация окулиста — Vis OD:OS=0,4:0,4. Глазное дно — без патологий. Коррекция зрения — (-3). Диагноз «Смешанный астигматизм, амблиопия». На осмотре у детского гинеколога патология со стороны гениталий не обнаружена. После проведенных параклинических обследований ребенок направлен на консультацию генетика и установлен клинический диагноз «Леопард синдром, синдром множественного лентиго».
Выводы. Фенотип ЛС чрезвычайно гетерогенный, клинические проявления варьируют от незначительной дисморфии лица и множественных лентиго до тяжелой формой гипертрофической кардиомиопатии, умственной отсталости и глухоты. Наличие множественных лентиго позволяет заподозрить синдром и своевременно провести тщательное клиническое и генетическое обследование. Отсутствие лентиго в раннем детстве и сходство клинических проявлений с синдромом Нунана затрудняет раннюю диагностику синдрома. Чаще всего у детей с ЛС диагностируется гипертрофическая кардиомиопатия, которая должна быть под тщательным контролем кардиолога с целью предупреждения внезапной сердечной смерти.
Исследование выполнено в соответствии с принципами Хельсинской Декларации. Протокол исследования одобрен Локальным этическим комитетом всех участвующих учреждений. На проведение исследований получено информированное согласие родителей ребенка.
Авторы заявляют об отсутствии конфликта интересов.
Ключевые слова: Леопард синдром, лентиго, гипертелоризм глаз, гипертрофическая кардиомиопатия, PTPN11 ген.
ЛИТЕРАТУРА
1. Gorlin RJ, Anderson RC, Moller JH. (1971). The Leopard (multiple lentigines) syndrome revisited. Birth Defects Orig Artic Ser. 7 (4): 110–115.
2. Sarkozy A, Digilio MC, Dallapiccola B. (2008). Leopard syndrome. Orphanet J Rare Dis. 3: 13. https://doi.org/10.1186/1750-1172-3-13; PMid:18505544 PMCid:PMC2467408
3. Morteza Moatamedi A, Mohammad Derakhshan. (2019). Leopard syndrome: a case report and literature review. Clinical Medicine. 19 (3): s23. https://doi.org/10.7861/clinmedicine.19-3-s23; PMid:31345866 PMCid:PMC6752376
4. Вentires-Alj M, Kontaridis MI, Neel BG (2006). Stops along the RAS pathway in human genetic disease. Nat Med. 12: 283–285. https://doi.org/10.1038/nm0306-283; PMid:16520774.
5. Limongelli G, Pacileo G, Marino B, Digilio MC, Sarkozy A, Elliott P, Versacci P, Calabro P, De Zorzi A, Di Salvo G, Syrris P, Patton M, McKenna WJ, Dallapiccola B, Calabro R. (2007). Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 100: 736-741. https://doi.org/10.1016/j.amjcard.2007.03.093; PMid:17697839.
6. Sarkozy A, Conti E, Digilio MC, Marino B, Morini E, Pacileo G, Wilson M, Calabro R, Pizzuti A, Dallapiccola B (2004). Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 41: e68. https://doi.org/10.1136/jmg.2003.013466; PMid:15121796 PMCid:PMC1735759.
7. Limongelli G, Sarkozy A, Pacileo G, Calabro P, Digilio MC, Maddaloni V, Gagliardi G, Di Salvo G, Iacomino M, Marino B, Dallapiccola B, Calabro R (2008). Genotype-phenotype analysis and natural history of left ventricular hypertrophy in LEOPARD syndrome. Am J Med Genet A. 46: 620–628. https://doi.org/10.1002/ajmg.a.32206; PMid:18241070
8. Gorlin JR, Cohen MM, Levn LS: Leopard syndrome. Syndromes of the head and neck. Edited by: Gorlin JR, Cohen MM, Levn LS. (1990). New York: Oxford University Press: 461-464.
9. Ucar C, Calyskan U, Martini S, Heinritz W (2007). Acute myelomonocytic leukemia in a boy with LEOPARD syndrome (PTPN11 gene mutation positive. J Pediatr Hematol Oncol. 28: 123–125. https://doi.org/10.1097/01.mph.0000199590.21797.0b; PMid:16679933
10. Yu Nakagama, Ryo Inuzuka, Kayoko Ichimura, Munetoshi Hinata, Hiroki Takehara, Norihiko Takedo. (2018). Accelarated Cardiomyocyte proliferation in the heart of a neonate with LEOPARD Syndrome-associated Fatal Cardiomyopathy. Circulation: Heart Failure. 11: 1–3. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004660; PMid:29602897
Синдром LEOPARD. Случай редкого наследственного заболевания в практике врача-генетика | Хлевная
https://doi.org/10.1234/XXXX-XXXX-2016-8-40-42Аннотация
Рассмотрен случай редкого наследственного заболевания синдром LEOPARD у представителей четырех поколений семьи. Диагноз был поставлен по совокупности клинических и фенотипических признаков, подтвержден молекулярно-генетическими методами.
Ключевые слова
Об авторах
Л. А. Хлевная Республиканский специализированный центр медицинской генетики и пренатальной диагностики Министерства здравоохранения Донецкой Народной Республики
Россия
Т. В. Лысенко
Республиканский специализированный центр медицинской генетики и пренатальной диагностики Министерства здравоохранения Донецкой Народной Республики
Россия
Н. В. Мазурик
Республиканский специализированный центр медицинской генетики и пренатальной диагностики Министерства здравоохранения Донецкой Народной Республики
Россия
Список литературы
1. Джонс Кеннет Л. Наследственные синдромы по Дэвиду Смиту. Атлас-справочник. М.: Практика, 2011. - 1024 с.
2. Gorlin R.J., Anderson R.C., Blaw M. Multiple lentigines syndrome // Am. J. Dis. Child. - 1969. - Vol. 117. №6. - P. 652-662.
3. Digilio M.C., Conti E., Sarkozy A. et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene // Am. J. Hum. Genet. - 2002. - Vol. 71. №2. Р.389-394.
4. Kalidas K., Shaw A.C., Crosby A.H. et al. Genetic heterogeneity in LEOPARD syndrome: two families with no mutations in PTPN11 // J. Hum. Genet. - 2005. - Vol. 50. №1. Р. 21-25.
5. Legius E., Schrander-Stumpel C., Schollen E.J. et al. PTPN11 mutations in LEOPARD syndrome // Med. Genet. - 2002. - Vol. 39. №8. - Р. 571-574.
6. Ogata T, Yoshida R. PTPN11 mutations and genotype-phenotype correlations in Noonan and LEOPARD syndromes // Pediatr. Endocrinol. Rev. - 2005. - Vol. 2. №4. - Р. 669-674.
Для цитирования:
Хлевная Л.А., Лысенко Т.В., Мазурик Н.В. Синдром LEOPARD. Случай редкого наследственного заболевания в практике врача-генетика. Медицинская генетика. 2016;15(8):40-42. https://doi.org/10.1234/XXXX-XXXX-2016-8-40-42
For citation:
Khlevnaya L.A., Lisenko T.V., Mazurik N.V. The LEOPARD syndrome. Тhe case of a rare hereditary disease in the practice of physician-geneticist. Medical Genetics. 2016;15(8):40-42. (In Russ.) https://doi.org/10.1234/XXXX-XXXX-2016-8-40-42
Просмотров: 461
ОрфаМир
- Синонимы:
- Cardiomyopathic lentiginosis, Familial multiple lentigines syndrome
- Код МКБ-10:
- Q87.8
- Orpha №
- ORPHA500
- MIM №
- 151100, 611554
Описание и характеристика. СЛ является редким заболеванием, которое характеризуется множественными аномалиями, главным образом они касаются кожных покровов, лица и сердца. В клинической картине обращает на себя внимание генерализованный лентикулярный меланоз, нарушение проводимости сердца. У этих больных лицевой дисморфизм представлен такими признаками, как широкое расположение глазниц (гипертелоризм), стенозом легочной артерии, абнормальным развитием гениталий, замедлением физического развития и сенсорнонейральной тугоухостью.
Распространённость и наследование. <1 / 1 000 000, аутосомно-доминантный тип наследования. В настоящее время в литературе описано 200 больных с СЛ. Однако, распространенность заболевания не установлена. Генетический анализ установил мутацию гена RAF1.
Клиника, диагностика и лечение. В диагностическом процессе большое значение придается выявлению кожных проявлений: лентикулярный меланоз, который и побуждает к активному поиску множественных аномалий ребенка. Дифференциальный диагноз проводится с нейрофибрамотозом (синдром Noonan). Специальных методов лечения не существует. Рекомендуется ежегодно проводить исследования слуха.
Link to Orphanet
Эксперты и учреждения, специализирующиеся в группе МКБ-10: Q00-Q99 Врожденные аномалии [пороки развития], деформации и хромосомные нарушенияСиндром леопарда
Я тоже участвовал в становлении России как крупнейшего зернового экспортера. В какой-то момент мне показалось, что я свою задачу выполнил, и возник вопрос: что же дальше? Я уже был настоящим госслужащим-«мамонтом». Все, кто начинал вместе со мной, ушли на покой или эмигрировали: получили американское гражданство и строят свою жизнь за океаном. Мой босс, американец, предлагал заманчивые варианты обустройства в любом городе США. Я мог там за несколько лет стать президентом крупной компании. Но такая перспектива меня не привлекала. Я гражданин России – по сути, по паспорту, по национальному характеру. Я люблю свою страну, мне в ней комфортно и хорошо жить. К тому же, когда я начинал, Америка была более патриархальной и человечной. А сейчас мой начальник, мормон и отец восьмерых детей, вынужден сидеть в кабинете с открытой дверью: боится, что кто-нибудь ворвется и обвинит его в изнасиловании. Сейчас подашь кому-нибудь пальто – могут обвинить в сексуальных домогательствах. Был случай, когда нашего чиновника потащили в полицию за то, что он попросил продавщицу примерить бюстгальтер для своей жены. Когда началась «эпоха политкорректности», когда стало необходимо соответствовать мейнстриму и говорить только то, что положено, я понял: это выше моих сил. Но и ситуация, когда за меня решают, куда ехать и что делать (неизбежная в системе крупной монополии), больше не устраивала. Бывает, что мужчина стремится поменять жизнь, вот как женщина иногда хочет изменить прическу. Раздражало, что приходится выполнять чужие указания. Я хотел сам определять свою судьбу. Но было страшно сниматься с насиженного места, панически боялся потерять то, ради чего столько трудился. Но все получилось, как с расторжением брака. Те, кто прошел через крушение своей семьи, понимают: они сначала тоже боялись поменять жизнь. Но сделали этот шаг. Когда я разводился, было очень тревожно, потому что жена полностью отгородила меня от быта. А потом переборол страх и стал жить один. Вот так же и с работой получилось. Вы просто хотите поменять свою жизнь, и когда этот момент наступает, точно знаете, что это он. Я полетел в отпуск на сафари. Утром подъехал на машине к какому-то болотцу, где было много-много птиц – цапель, журавлей, пеликанов, а рядом в зарослях прятался леопард. Я долго наблюдал за ним и вдруг сказал себе: «Любой ценой я хочу быть таким же, как эта кошка!» Энергетика этого места выбила меня из привычной колеи. Я вернулся и сразу подал прошение об отставке. И вот я уже не большой начальник, окруженный водителями и секретарями, а идущий по лезвию бритвы бизнесмен. Я стал продавать поездки в Африку, предполагая, что на этом направлении меньше конкуренция. Я был готов отправить своих клиентов в самые дикие и опасные племена, где можно нырять с крокодилами и делать то, что запрещено всем остальным. Как бывший военный офицер, служивший в горячих точках Черного континента, я понимал, насколько важна личная охрана и проверенный проводник, а также связи с местным правительством, разведкой и частными компаниями. И вот я отправляю группу в Ботсвану, сложнейший и очень дорогой тур. Все в путешествии продумано до мелочей, но в какой-то момент смотрю – нет шести паспортов в офисе. А люди уже заплатили колоссальные деньги. Через три дня вылет, а документов нет. Перерыл все дома, даже балкон, на который три года не выходил. Офис прошерстили с милицией – безрезультатно. Это был тяжелый момент. Сложно придумать что-нибудь хуже, чем потерять паспорта туристов. На этом бы моя молодая компания и закрылась. Но тут ночью раздается звонок от одного туриста, который сообщает, что его жена по непонятной причине положила шесть чужих паспортов в свою сумку. С тех пор документы хранятся у меня только в сейфе, да и вообще: деловая дисциплина считается не менее важной, чем умение договориться с вождями первобытных племен. В саванне своя философия. Там говорят: иногда ошибка может уберечь тебя от большого несчастья. А иногда то, что кажется твоей большой победой, на самом деле может быть крупнейшим поражением. Самое главное: не изменяй себе, и тогда тебе не изменит судьба. Если ты по натуре бродяга, не стоит искать себе дом – ты не будешь там счастлив. Кошелек мой стал тоньше, но сердце поет. С тех пор как уволился, ни разу не был в цивилизованных краях – Америке или Европе. Только Африка, только хардкор! Никогда не поздно осознать, что жизнь – одна, и переиграть ее полностью на свежих подмостках среди новых декораций.
Прогрессирующая гипертрофическая кардиомиопатия при синдроме LEOPARD - Хирургия. Журнал им. Н.И. Пирогова - 2012-12
Синдром LEOPARD - это редкое аутосомно-доминантное заболевание, которое является аллельным вариантом синдрома Нунан [2-5], и вызывается мутациями в генах PTPN11 [5, 8, 22], RAF1 [18, 35, 37] и BRAF [21, 38]. В литературе имеется около 200 описаний этого заболевания [40].
Название синдрома дано с учетом начальных букв английских обозначений основных симптомов синдрома, в соответствии с R. Gorlin и соавт. (1969) [15, 16]. Lentigo - лентиго (до 100%) [3, 7]. ECG - аномалии ЭКГ (75-80% из них 46% гипертрофия левого или обоих желудочков [69], в сочетании с зубцом Q (19%), удлиненным QTc (23%) и нарушением реполяризации (42%) [23]; в 23% наблюдений с нарушениями проводимости и в 19% с аномалиями зубца P. Ocular hypertelorism - глазной гипертелоризм (75%). Pulmonic stenosis - стеноз легочной артерии (95%). Abnormalities of genitalia - аномалии гениталий (50%), включающие агенезию или гипоплазию гонад, гипогонадизм. Retardation of growth - отставание в росте (40-50%) [14, 48].
В 50% наблюдений имеются крыловидные лопатки, в 35% - шейный птеригиум. Отмечается воронкообразная или килевидная деформация грудной клетки. Deafness - глухота (15-25%) [39]. Возможна умеренная умственная отсталость.
Приводим клиническое наблюдение.
Больной Е., 17 лет, поступил с диагнозом: врожденный порок сердца; гипертрофическая кардиомиопатия с обструкцией выносящего тракта левого желудочка; митральная недостаточность IV степени. Состояние после имплантации двухкамерной частотно-адаптивной кардиостимулирующей системы 11.04.05. Нарушения ритма сердца и проводимости: суправентрикулярная и желудочковая экстрасистолия. Недостаточность кровообращения III-IV функционального класса.
Из анамнеза: со слов матери, второй ребенок, от второго брака. Родился на 7-м месяце гестации. Роды стимулировали медикаментозно в связи с резко ослабленной родовой деятельностью. Масса ребенка 3500 г, рост 51 см. При рождении закричал сразу, отмечалась деформация грудной клетки, темно-коричневые пятна на коже нижних конечностей и распространенная отечность подкожной жировой клетчатки. Диагностирован врожденный порок сердца (идиопатический гипертрофический субаортальный стеноз). Наблюдался в НИИ педиатрии. С 6 лет появились лентиго на лице и по всему телу, усугубилась деформация грудной клетки, появилась деформация стоп нижних конечностей, отставал от сверстников в физическом развитии. С 9-летнего возраста стал отмечать значительное ухудшение состояния, снижение толерантности к нагрузкам, перебои в работе сердца, кратковременные эпизоды потери сознания. В возрасте 11 лет находился на обследовании и лечении в РНЦХ. 04.04.05 поставлен диагноз идиопатического гипертрофического субаортального стеноза, произведена имплантация двухкамерного электрокардиостимулятора (ЭКС) «Insigia I Ultra» с целью стабилизации сердечного ритма и профилактики синкопальных состояний.
11.08.10 выполнена реимплантация ЭКС в связи с истощением батареи ЭКС.
При настоящем обследовании: больной небольшого роста (160 см), астенического телосложения. На коже множественные лентиго, расположенные по всему телу, на ногах два пятна темно-коричневого цвета размером более 1,5 см, гиперэластичность кожи, акроцианоз нижних конечностей, несколько келоидных рубцов после оперативного вмешательства и несколько «папиросных» рубцов. Отмечаются лицевой дисморфизм, низкопосаженные уши, килевидная деформация грудной клетки, искривление позвоночника (кифосколиоз грудопоясничного отдела), гипермобильность суставов (рис. 1).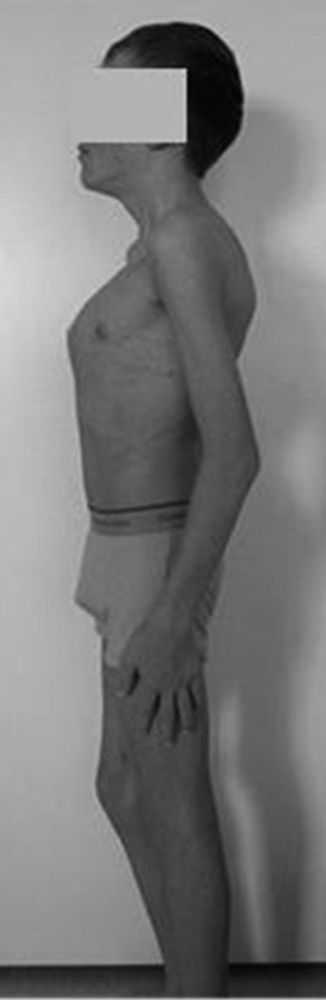 Рисунок 1. Внешний вид больного. Слух сохранен, интеллектуальное развитие соответствует возрасту, гипогенитализма нет.
Рисунок 1. Внешний вид больного. Слух сохранен, интеллектуальное развитие соответствует возрасту, гипогенитализма нет.
Аускультативно: дыхание в легких везикулярное, хрипов нет. ЧД 18 в 1 мин. Тоны сердца ритмичные, ясные, выслушивается систолический шум на верхушке. ЧСС 100 в 1 мин. АД 110/70 мм рт.ст. Живот мягкий, безболезненный. Печень и селезенка не увеличены. Стул в норме. Симптом поколачивания с обеих сторон отрицательный. Периферических отеков нет. Физиологические отправления в норме. Данный фенотип характерен для синдрома LEOPARD, с этим предварительным диагнозом выполнено полное клиническое и генетическое обследование с акцентом на выявление возможных внутрисердечных аномалий.
Результаты проведенного обследования
ЭКГ: синусовая тахикардия (ЧСС 100 в 1 мин), отклонение электрической оси сердца влево, нарушение и замедление внутрижелудочковой проводимости (интервал QRS 0,12 с). Гипертрофия правого желудочка. Гипертрофия и перегрузка левого желудочка. Изменения миокарда левого желудочка (ЛЖ).
Холтер-ЭКГ: в период мониторирования регистрировался ритм ЭКС в режиме стимуляции предсердий. Зафиксировано 162 эпизода нарушения чувствительности ЭКС. Средняя ЧСС 84 в 1 мин, максимальная ЧСС 154 в 1 мин, минимальная ЧСС 71 в 1 мин. Наджелудочковая эктопическая активность состояла из 25 сокращений, в том числе 1 эпизод групповой экстрасистолии из 3 комплексов. Желудочковая эктопическая активность состояла из 118 сокращений, из которых 1 парное - класс по Лауну 4А.
ЭхоКГ: выраженная концентрическая гипертрофия миокарда левого желудочка с обструкцией выходного тракта левого желудочка (ЛЖ), гипертрофированная переднелатеральная папиллярная мышца, прикрепляющаяся к апикальному сегменту боковой стенки ЛЖ. Глобальная и локальная систолическая функция левого желудочка не нарушена. Диастолическая дисфункция ЛЖ III типа. Митральная регургитация IV степени (отрыв хорд задней створки). Кровоток в выходном тракте ЛЖ: Vmax=5,4 м/с PG=117/61 мм рт.ст. (рис. 2).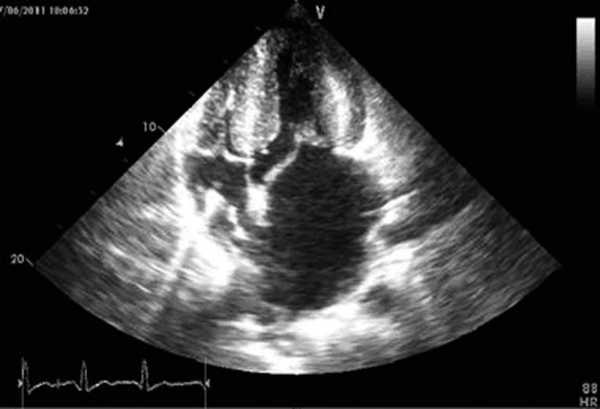 Рисунок 2. Трансторакальная эхокардиография до операции. а - систолическая фаза.
Рисунок 2. Трансторакальная эхокардиография до операции. а - систолическая фаза.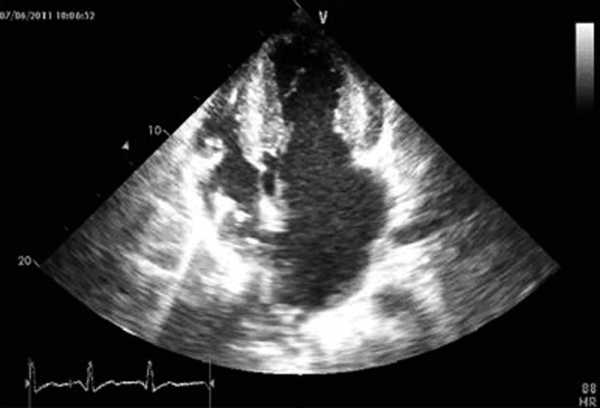 Рисунок 2. Трансторакальная эхокардиография до операции. б - диастолическая фаза.
Рисунок 2. Трансторакальная эхокардиография до операции. б - диастолическая фаза.
Полипозиционная рентгенография и рентгеноскопия органов грудной клетки: венозный тип сердечного застоя в малом круге кровообращения. Корни легких малоструктурны, расширены. Увеличение обоих предсердий. Функциональные признаки митральной недостаточности.
Коронароангиография: сбалансированный тип кровоснабжения сердца. Коронарные артерии не изменены.
Компьютерная томография: выраженная гипертрофия миокарда ЛЖ: толщина межжелудочковой перегородки (МЖП) - переднесептальный 2,6 см, нижнесептальный 2,5 см. Толщина миокарда ЛЖ переднебоковой стенки - 2,3 см, нижнебоковой стенки 2,3 см, в апикальной части 0,97 см. Выходной тракт ЛЖ 18 мм.
Генетическое исследование: была проведена ДНК-диагностика у членов семьи (пробанд и родители) с целью подтверждения диагноза и установления происхождения мутации. Методом прямого секвенирования по Сенгеру были проанализированы кодирующие и регуляторные последовательности генов PTPN11, RAF1 и BRAF. В гене PTPN11, ответственном за 80% всех известных наблюдений синдрома LEOPARD, была выявлена мутация p.Thr468Met в гетерозиготном состоянии [24]. В образцах ДНК клинически здоровых родителей пациента мутации не выявлено. Таким образом, было установлено происхождение мутации de novo и отсутствие риска повторного рождения ребенка с этим заболеванием в семье.
На основании проведенного обследования уточнен анатомический диагноз: первичная распространенная гипертрофическая кардиомиопатия (гипертрофия папиллярной мышцы, средней части выносящего тракта МЖП), фиброзная субаортальная мембрана, дисплазия митрального клапана с отрывом хорд задней створки.
Лечение
Первый этап - 18.07.11 выполнена операция: расширенная миоэктомия (продольная редуцирующая объем резекция папиллярных мышц, миоэктомия перегородки), резекция субаортальной мембраны, универсальное хордосохраняющее протезирование митрального клапана механическим протезом «OptiForm-27» в условиях искусственного кровообращения и холодовой кардиоплегии (рис. 3).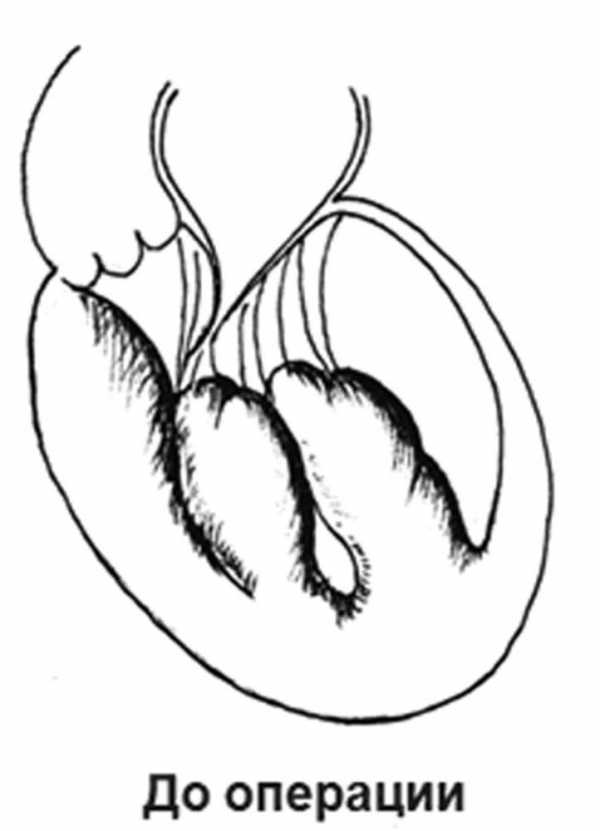 Рисунок 3. Схема операции. а - вид до операции.
Рисунок 3. Схема операции. а - вид до операции.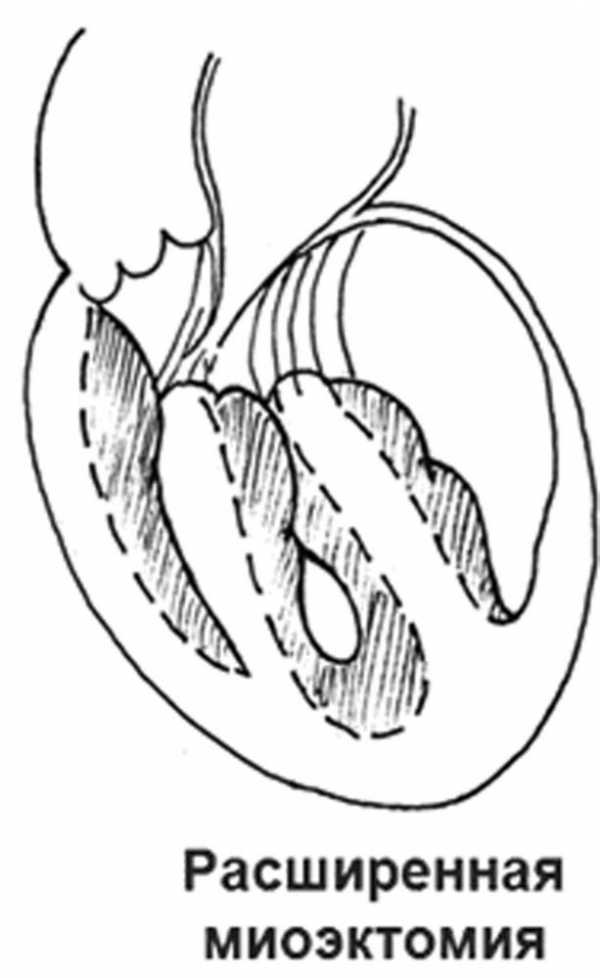 Рисунок 3. Схема операции. б - расширенная миоэктомия.
Рисунок 3. Схема операции. б - расширенная миоэктомия.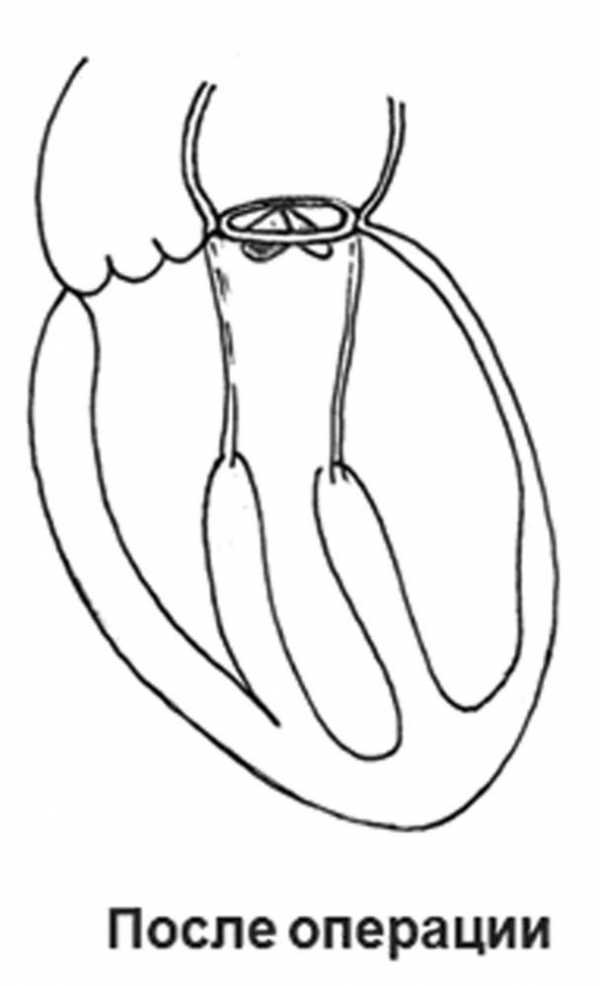 Рисунок 3. Схема операции. в - вид после универсального хордосохраняющего протезирования митрального клапана механическим протезом.
Рисунок 3. Схема операции. в - вид после универсального хордосохраняющего протезирования митрального клапана механическим протезом.
Второй этап - 02.12 выполнена имплантация кардиодефибриллятора (ИКД).
Патогистологическое исследование операционного материала: признаки дисплазии соединительной ткани створок митрального клапана с очагами склероза (рис. 4).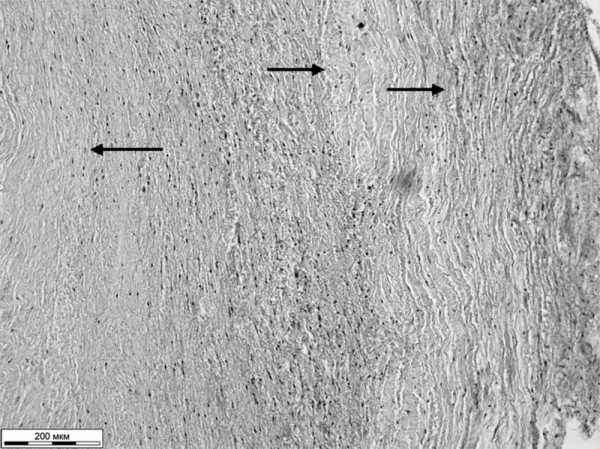 Рисунок 4. Микрофотография. а - митральный клапан с дисплазией соединительной ткани. Окраска гематоксилином и эозином. Ув. 10.
Рисунок 4. Микрофотография. а - митральный клапан с дисплазией соединительной ткани. Окраска гематоксилином и эозином. Ув. 10.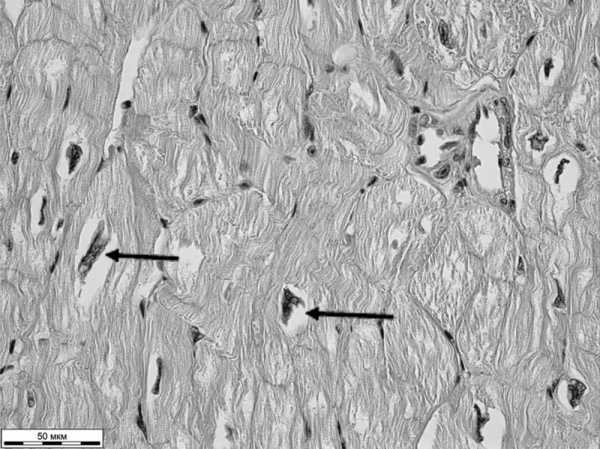 Рисунок 4. Микрофотография. б - межжелудочковая перегородка. Зазубренность ядер и просветления. Окраска гематоксилином и эозином. Ув. 40.
Рисунок 4. Микрофотография. б - межжелудочковая перегородка. Зазубренность ядер и просветления. Окраска гематоксилином и эозином. Ув. 40.
В миокарде межжелудочковой перегородки признаки гипертрофической кардиомиопатии: кардиосклероз, неупорядоченность расположения волокон, гипертрофия кардиомиоцитов, зазубренность контуров ядер и просветление вокруг них. Стенки мелких интрамуральных артерий утолщены за счет гипертрофии среднего слоя (рис. 5).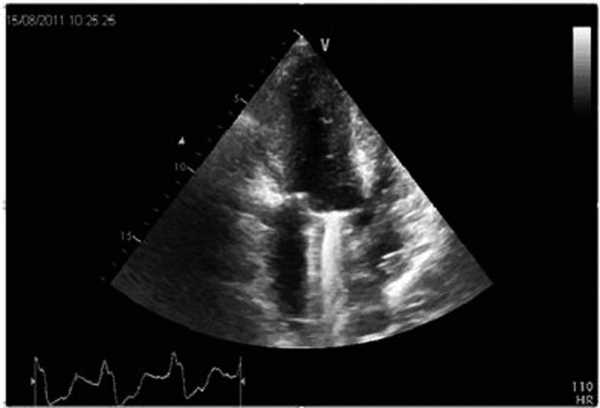 Рисунок 5. Трансторакальная эхокардиография после операции. а - систолическая фаза.
Рисунок 5. Трансторакальная эхокардиография после операции. а - систолическая фаза.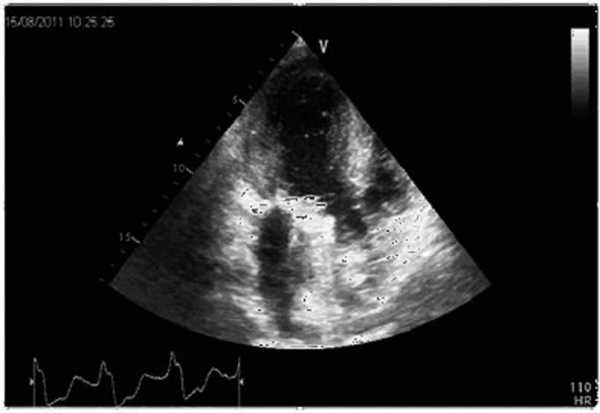 Рисунок 5. Трансторакальная эхокардиография после операции. б - диастолическая фаза.
Рисунок 5. Трансторакальная эхокардиография после операции. б - диастолическая фаза.
Ранний послеоперационный период протекал с умеренной сердечно-сосудистой и печеночно-почечной недостаточностью, гипертермией, гиперферментемией, полисерозитом. Через 3-6-10 мес состояние больного относительно удовлетворительное. Клинических признаков недостаточности кровообращения, гемодинамически значимых нарушений ритма сердца, синкопе не наблюдается. Пациент отмечает улучшение общего самочувствия в виде регресса одышки и перебоев в работе сердца, повышения толерантности к физическим нагрузкам.
Результаты контрольных исследований после операции
ЭхоКГ: отмечается положительная динамика в виде увеличения КДО ЛЖ с 77 до 116 мл, увеличения УО с 58 до 83 мл и значительного уменьшения скорости кровотока и градиента давления в выходном тракте ЛЖ: Vmax с 5,4 до 1,2 м/с, PGr/MGr со 117/61 до 6,0/2,9 мм рт.ст. (рис. 6).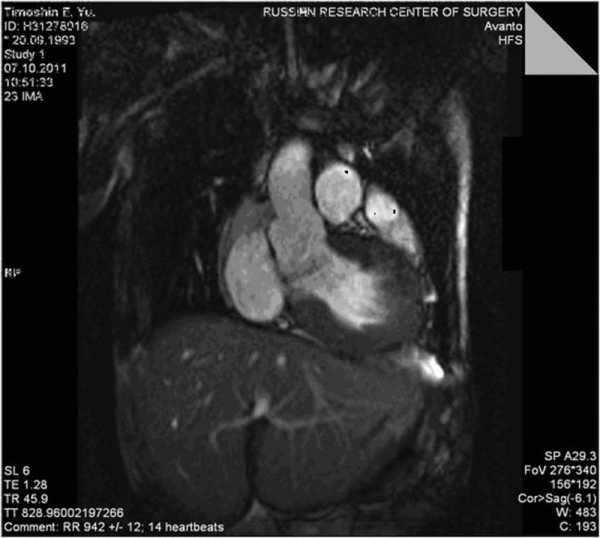 Рисунок 6. МРТ сердца после операции. а - систолическая фаза.
Рисунок 6. МРТ сердца после операции. а - систолическая фаза.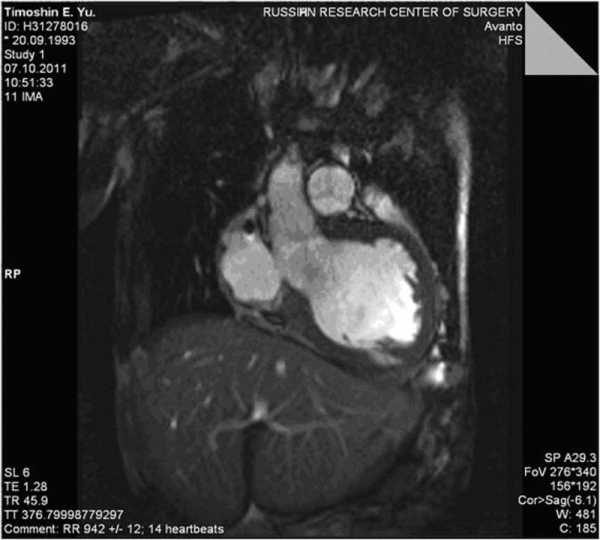 Рисунок 6. МРТ сердца после операции. б - диастолическая фаза.
Рисунок 6. МРТ сердца после операции. б - диастолическая фаза.
МРТ сердца: толщина миокарда ЛЖ: МЖП 2,1 см, ЗСЛЖ 1,1 см, боковая стенка ЛЖ 1,4 см. В проекции левого атриовентрикулярного отверстия определяются структуры клапанного протеза (рис. 7).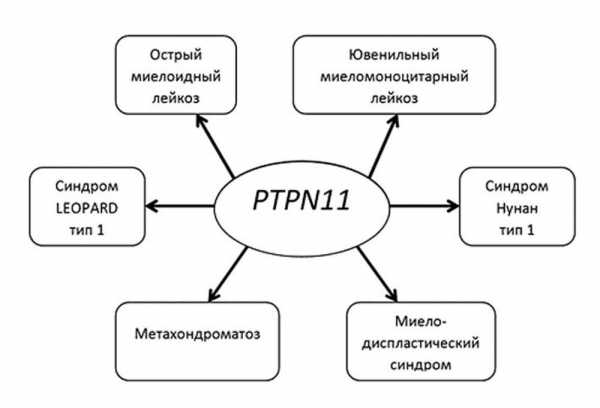 Рисунок 7. Аллельная серия заболеваний, вызываемых мутациями в гене PTPN11.
Рисунок 7. Аллельная серия заболеваний, вызываемых мутациями в гене PTPN11.
Несмотря на специфичность клинической картины синдрома LEOPARD, диагностика этого заболевания затруднена в связи с недостаточной осведомленностью врачей о редких наследственных синдромах. Наличие у больных множественных лентиго позволяет заподозрить синдром LEOPARD, что требует проведения клинического и генетического исследования для верификации диагноза. У этих больных часты сочетанные пороки клапанов и сердечной мышцы, такие как стеноз легочной артерии, дефект межпредсердной перегородки, субаортальный стеноз и гипертрофическая кардиомиопатия. Больным требуется проведение комплекса обследований сердечно-сосудистой системы и поэтапного хирургического лечения.
Одним из наиболее эффективных методов лечения обструктивной формы ГКМП является хирургическое вмешательство [25, 26]. Двумя основными общепринятыми хирургическими способами лечения ГКМП являются септальная миоэктомия [27, 28] и алкогольная септальная аблация [29, 30]. Успех последней зависит от вариабельности анатомии ветви септального перфоранта, что в 20-25% наблюдений не позволяет достичь необходимого эффекта [31]. При распространенной форме гипертрофической кардиомиопатии и тем более при сочетании с другими внутрисердечными патологическими изменениями, единственный способ лечения - операция в условиях искусственного кровообращения [32].
Считаем, что при распространенной форме гипертрофической кардиомиопатии, когда в процесс вовлечены все стенки и папиллярные мышцы, операция эффективна лишь при выполнении расширенной септальной миоэктомии, резекции или полном удалении папиллярных мышц [33-36]. Только такая процедура способна увеличить КДО ЛЖ и уменьшить выраженность его диастолической дисфункции.
В настоящем клиническом наблюдении еще до операции выявлена выраженная дисплазия митрального клапана (клапан Барлоу), что нашло подтверждение при визуальном осмотре во время операции (избыточная площадь створок с отрывом хорд к задней створке). Это в данной ситуации является веским показанием к протезированию, гистологическое изучение створок после операции подтвердило диагноз [37, 38].
Поскольку при ГКМП высока вероятность желудочковых тахиаритмий и внезапной сердечной смерти, во всех таких наблюдениях должна быть проведена стратификация риска внезапной сердечной смерти на начальных этапах обследования и выявлены эпизоды тахиаритмий, а также необъясненные синкопе в анамнезе [39-50]. Имплантируемые кардиодефибриллирующие системы являются эффективным способом превентивной терапии угрожающих жизни нарушений ритма и предотвращения возможной внезапной сердечной смерти [51], в связи с чем в описанном выше наблюдении завершающим этапом хирургического лечения стала имплантация кардиодефибриллятора.
Особенностью естественного течения синдрома LEOPARD является повышенный по сравнению с популяционным риск развития неоплазий. Гены, ответственные за развитие синдрома LEOPARD, относятся к семейству протоонкогенов, и мутации в них ассоциированы с риском развития лимфопролиферативных заболеваний. Например, разные мутации в гене PTPN11 могут быть причиной аллельной серии заболеваний с различным клиническим фенотипом, но повышенным риском онкологических заболеваний. Поэтому важной частью катамнестического наблюдения за больными с синдромом LEOPARD является плановый контроль клеточного состава крови и консультации онколога ежегодно.
Таким образом, представленное описание редкого наблюдения свидетельствует о необходимости точной этиологической диагностики различных форм кардиомиопатий. «Золотым стандартом» верификации наследственных синдромов является прямая ДНК-диагностика. Только при наличии подтвержденного диагноза можно персонализированно подойти к лечению и прогнозу.
Объем хирургического вмешательства должен быть полноценным. Необходимо устранить патологическое изменение клапана, максимально увеличить объем полости левого желудочка и предотвратить угрожающие жизни аритмии.
Выявление генетической причины заболевания позволяет проводить быстрое тестирование родственников, в том числе выявление малосимптомных форм, пренатальную диагностику, осуществлять медико-генетическое консультирование.
Ген PTPN11 (синдром LEOPARD), выявление мутаций в экзонах 7, 12, 13
Подготовка к исследованию: Специальной подготовки к исследованию не требуется. Исследуемый материал: Взятие крови
Синдром LEOPARD
- очень редкое наследственное заболевание. В современной медицинской литературе описано всего немногим более сотни случаев. Название представляет собой аббревиатуру, составленную для удобства запоминания из первых букв английских названий симптомов. Кроме того, самый часто встречающийся симптом (84%) – лентиго-пигментарные пятна разных оттенков коричневого цвета делают кожу больного похожей на шкуру леопарда. Кроме этого, есть еще несколько легко заметных симптомов: широко расставленные глаза, задержка физического роста и развития и аномалии развития половых органов у мужчин. Заметить аномалии развития половых органов у женщин сложнее. Проявления со стороны сердечно-сосудистой системы выражаются в изменениях ЭКГ и сужении легочной артерии. Кроме того, у некоторых больных обнаруживаются сужение аорты и пролапс митрального клапана. Из-за аномалий развития слухового нерва может возникать глухота.
Причиной заболевания, как правило, является одна из пяти мутаций в гене PTPN11, кодирующем белок SHP2. SHP2 – белковая тирозинфосфатаза – сигнальная молекула, регулирующая процессы клеточного роста, деления и дифференцировки. Заболевание наследуется аутосомно-доминантно. Это значит, что мутации в одной из двух копий гена на одной из хромосом достаточно для развития заболевания. У больного с синдромом LEOPARD с вероятностью 50% будут рождаться дети с тем же заболеванием.
Мутации, вызывающие заболевание, могут находиться в 7, 12 или 13 экзонах. Их поиск осуществляется методом прямого секвенирования. Может проводиться пренатальная диагностика.
Выявление мутаций проводится путем прямого автоматического секвенирования. Секвенирование – это метод, позволяющий «прочитать» последовательность ДНК целиком, то есть узнать, какой именно нуклеотид находится в ней на каком месте. Секвенирование производится с помощью полимеразной цепной реакции. Используя специальные реактивы, можно получать копии фрагментов ДНК разной длины, один конец которых находится в строго определенном месте, а другой отстоит от него на произвольное число нуклеотидов. При этом будет точно известно, какой из четырех нуклеотидов занимает последнюю позицию в синтезированном фрагменте. Сравнив длины полученных фрагментов, можно восстановить всю последовательность. На сегодняшний день эта процедура может быть полностью автоматизирована.
С помощью генетического анализа можно подтвердить диагноз или провести пренатальную диагностику.
Синдром леопарда - Все сразу — LiveJournal
Свой рассказ я начну с клинического наблюдения. Одному моему пациенту, известному (в узких кругах) и образованному (в широком смысле) человеку, заменили один из клапанов сердца. Были все показания, перед операцией мы неоднократно обсуждали потенциальные риски и предстоящую пожизненную антикоагулянтную терапию. Слово «антикоагулянтный» можно легко и без потери смысла заменить на «кроверазжижающий».Дело в том, что механический протез клапана, хоть и сделан из «правильных» материалов, но все-таки немножко чужеродный. Если на дискотеку села Большие Грязи придет афроамериканец с компанией друзей из соседнего села, все мы знаем, что первым начнут бить чужестранца, как бы он ни был ассимилирован. А если ты чужеродный протез клапана, то к тебе тоже начинают приставать – тромбоциты.
К тебе пристают тромбоциты, которые зовут других тромбоцитов,
и вот уже ты облеплен тромбоцитами со всех сторон.
На поверхности протеза начнет расти тромб.
Именно поэтому пациентам после протезирования клапана необходимо пожизненно принимать кроверазжижающие препараты. Например, варфарин, про который я тут рассказывал месяц назад. Как я уже говорил, варфарин должен поддерживать определенную вязкость крови, при которой протез клапана не будет тромбироваться, а риск кровотечений будет минимален. Атос, Портос и Компромисс. Для этого врачи и ученые придумали специальный показатель вязкости крови – МНО (в данном случае показатели МНО должны были укладываться в диапазон от 2,5 до 3,5), а этот МНО не так просто подобрать…
Мы с пациентом остановились на двух с половиной таблетках варфарина, которые позволяли нам достичь целевых значений.
И вот звонит пациент и сообщает, что у него появились синяки на теле. Я предлагаю ему срочно приехать и вижу перед собой, натурально, человека-леопарда. Хорошо, что это просто синяки, а не геморрагический инсульт или мощное желудочное кровотечение.
Во время осмотра я вижу на груди в проекции сердца специфические следы,
похожие на маленькие значки марки мерседес-бенц.
Гирудин, вещество, выделяемое слюнными железами пиявок, является прямым ингибитором тромбина (одного из факторов свертывания). Именно гирудин сделал кровь еще более жидкой и сместил то хрупкое равновесие, которого мы добивались с таким трудом. На вопрос о причинах суицидального поведения пациент ответил, что он просто хотел немножечко помочь своему, без сомнения, многострадальному сердцу методами народной медицины. По счастью все закончилось легкой леопардовостью кожных покровов пациента.

А вот другой пример. Ни одно лекарственное вещество в кардиологической практике не вызывает у пациентов такой негативной реакции, как статины (лекарства для снижения уровня «плохого» холестерина и воспаления стенок артерий, которые минимизируют риски смерти от всех причин на 30% и на 42% снижают риск смерти от ишемической болезни сердца)
Я теряюсь в догадках о причине этого явления. Пациенты с порога заявляют:
- У меня высокий холестерин, но статины я пить не буду.
Другие приносят статьи из бульварных газет, в которых известный доктор естественных наук, выводит ядовитые статины на чистую воду.
Пациенты очень хотят снизить холестерин,
но при этом сохранить печень, как они говорят.
Я, кстати, давно понял, печень – главный орган россиянина.
Ради этих целей они готовы тоннами есть чеснок, не обращая внимания на социальные последствия этого пристрастия и полную бесполезность чеснока и его препаратов для снижения холестерина.
А теперь еще новая сенсация потрясла моих пациентов. Красный дрожжевой рис! Он же содержит природный ловастатин. Они готовы платить за него «три счетчика», лишь бы природный. Никого не смущает тот факт, что ловастатин давно не используется в медицине по причине малой эффективности и высокой токсичности, а его доза в этом рисе может быть самой различной от нуля до смертельной.
Я пишу это не для того, чтобы посмеяться над своими пациентами. Я и сам полон предрассудков и заблуждений в вопросах, по которым не имею специального образования.
Однако частота бытового употребления слова «химия» с негативными коннотациями обратно пропорциональна успеваемости по этой самой химии в школе. Не нужно думать, что все лекарственные средства убивают вашу многострадальную печень, а все «природные» аналоги нежно гладят ее по глиссоновой капсуле и поют ей колыбельные.
Если врач назначает вам препарат, чтобы продлить вашу жизнь –
принимайте и не ищите «безопасных» аналогов в природе.
Неоспоримым преимуществом лекарственных препаратов является их точная дозировка, четкие показания и противопоказания, особенности лекарственного взаимодействия, известный лечебный и предсказуемый побочный эффекты. (Лекарственными препаратами я называю вещества, доказавшие свою эффективность и безопасность в многоцентровых, плацебоконтролируемых исследованиях).
Уверен, что не существует берестяных грамот с подробным описанием показаний, противопоказаний, побочных эффектов, фармакокинетики и фармакодинамики лекарственных смесей, которые применялись на Руси. И это понятно.
«Старый» - не значит «умный».
Классическая медицина развивалась и видоизменилась вслед за развитием современной науки. Например, без изобретения микроскопа невозможно себе представить открытие микроорганизмов, которое в свою очередь послужило изобретению антибиотиков.
Тактики лечения острого инфаркта миокарда сегодня и 15-20 лет назад отличаются, как коньяк «командирский» от манной каши.
Я сам никогда не назначу лекарственный препарат без необходимости и всегда настаиваю прежде всего на изменении образа жизни – регулярных аэробных нагрузках, изменении стереотипа питания и отказе от вредных привычек.
Конечно, современная медицина далека от совершенства. Но лечение, основанное на принципах доказательной медицины, – это то, что мы, врачи, можем предложить нашим пациентам уже сейчас.
Да прибудет с вами Сила!
Стволовые клетки с дефектом помогут раскрыть тайны «синдрома леопарда»
Стволовые клетки с дефектом - это не результат халатности, а ценный материал для исследователей. Источник: AP
Впервые из стволовых клеток были получены клетки миокарда с серьезным дефектом. Именно клеток, из которых состоит пораженная кардиомиопатией сердечная мышца, не хватало исследователям для того, чтобы больше узнать об этом заболевании.
Обычно стволовые клетки ассоциируются с регенеративной медициной и восстановлением органов. Их уже применяют при лечении лейкоза, а список экспериментальных методик намного шире: от исправления дефектов шейки бедра до выращивания новых зубов. Путем целенаправленного перепрограммирования из стволовых клеток можно получить практически любую ткань, так как именно из стволовых клеток формируется растущий организм в ходе эмбрионального развития.
Клетки, образуемые при делении яйцеклетки, вначале универсальны, а со временем приобретают более узкую специализацию. Источник: http://ru.wikipedia.org
То, что иногда полученные из стволовых клеток ткани отличаются по своим свойствам от нормальных, в подавляющем большинстве случаев является фундаментальным недостатком, который ставит крест на дальнейшем использовании метода в медицине.
Но на этот раз группа исследователей из медицинского центра Маунт-Синай в США специально добивалось получения пораженных клеток.
Кардиомиопатия
Кардиомиопатия— это целый класс расстройств, связанных с поражением сердечной мышцы и грозящих тяжелыми последствиями, вплоть до смертельного исхода. Их могут вызывать самые разные причины от болезней, связанных с нарушением обмена веществ до врожденных пороков сердца. Именно врожденные нарушения развития, а еще точнее— болезнь, известная как синдром множественных лентигиний или просто синдром леопарда,— и интересовали американских медиков.
Синдром леопарда назван так по наиболее бросающемуся в глаза признаку— темным пятнам на коже. Но кроме пигментных пятен, низко посаженных ушей и большего расстояния между глазами синдром леопарда часто проявляет себя поражением миокарда, кардиомиопатией. Почему?
В поисках ответа на этот вопрос, и попутно рассчитывая на лучшее понимание развития кардиомиопатии вообще, кардиологи решили провести эксперимент со стволовыми клетками.
Что делали?
Медики вначале взяли у пациентов, больных синдромом леопарда, клетки кожи. Потом получили из них так называемые плюрипотентные стволовые клетки— те, которые могут превратится в любой другой вид клеток. И уже из них получили клетки миокарда, которые можно размножать in vitro, «в пробирке».
Клетки, растущие вне организма, можно подробно изучать, проверять на них действие лекарств и все это— без какого-то вмешательства в организм пациента. Ученым уже известно, что при синдроме леопарда нарушается работа некоторых ответственных за передачу сигналов внутри клетки белков (в частности работа так называемого RAS-белка), но теперь у них есть уникальная возможность узнать о заболевании больше в опытах на реальных живых клетках.
По словам исследователей, схожий подход может позволить в будущем получать культуры больных клеток от людей с разными редкими врожденными заболеваниями, а это, в свою очередь, сделает возможным подбор методов лечения.
Знание конкретных генов, которые «выходят из строя» при тех или иных болезнях так же может привести к их модификации: специально сконструированными вирусами уже удавалось вставить правильную копию гена в ДНК клетки и за счет этого значительно улучшить состояние больного.
gzt.ru
Онкогенетические синдромы кожи и ее придатков
Наследственные синдромы с поражением кожи (генодерматозы), составляют едва ли не самую многочисленную группу среди онкогенетических синдромов.Пигментная ксеродерма
Наследуется по аутосомно-рецессивному типу. Первичный дефект связан с нарушением процесса репарации ДНК от повреждения УФ-лучами и развиваются «болезни репарации ДНК». Этот термин появился после того, как было обнаружено, что в основе некоторых наследственных заболеваний лежит неспособность репарировать (восстанавливать исходную структуру) ДНК от повреждений, вызываемых внешними воздействиями.Впервые дефект репарации ДНК от повреждений, вызываемых УФ-лучами у человека, был обнаружен Кливером (1969) при пигментной ксеродерме. В дальнейшем получены данные о нарушении репарации и репликации ДНК еще при нескольких наследственных заболеваниях.
Симптоматика синдрома связана с солнечным облучением При рождении кожа имеет нормальный вид. Первые симптомы появляются примерно в возрасте 3 лет и прогрессируют в течение жизни. На участках кожи, наиболее подвергающихся солнечному воздействию (лицо, кисти рук), появляются разного размера, формы и цвета многочисленные веснушки, телеангиэктазии, обесцвеченные пятна, рубцы, участки атрофии и кератоза.
Часто поражаются веки и роговица, приводящие к слепоте. При прогрессировании кожных поражений, как правило, наступает злокачественное перерождение. Рак кожи развивается в молодом возрасте, часто бывает множественным и рано (до 30 лет) приводит к гибели. Кроме базальноклеточных и плоскоклеточных карцином кожи у больных встречаются меланомы, ангиомы и саркомы. Риск заболеть раком повышен в 1000 раз.
Полная изоляция от солнечного света предотвращает кожные поражения, поэтому больным рекомендуется вести ночной образ жизни. Лечение кожных нарушений симптоматическое.
При лечении новообразований не рекомендуется радиотерапия и применение химиопрепаратов. Гетерозиготные носители гена составляют группу повышенного онкологического риска. Вопрос о диспансеризации родственников больного решается на основании генеалогического анализа.
Синдром базальноклеточного невуса
Синдром базальноклеточного невуса (баэальноклеточная карцинома, синдром Горлина-Гольца) наследуется по аутосомно-доминантному типу, считается редким (около 1% всех множественных новообразований кожи) и входит в группу факоматозов.Главное проявление синдрома — множественные базальноклеточные карциномы, локализующиеся на лице, шее, спине и груди. Возраст, в котором возникают опухоли, сильно варьирует (от подросткового до 50 лет), но более молодой по сравнению со средним возрастом появления спорадических форм базалиомы.
У 65-75% больных развиваются одонтогенные кисты главным образом нижней челюсти. Синдром проявляется также рядом черепно-лицевых и скелетных аномалий: большая голова, гипертелоризм, фронтальные и теменные шишки, умеренный прогнатизм, spina bifida, сколиоз, кифоз, укороченные метакарпальные кости, раздвоение ребер, синдактилия, субкортикальные кистозные изменения трубчатых костей, аномалии турецкого седла и др. Возможна марфаноподобность.
Интеллект часто снижен вплоть до умственной отсталости. Поскольку имеются данные о повышенной чувствительности клеток таких больных к ионизирующему облучению, мерой профилактики новообразований является строгое ограничение рентгенодиагностических обследований, радиотерапевтических процедур и применения радиомиметических агентов. Желательно полное исключение таких воздействий.
Синдром диспластического невуса
Синдром диспластического невуса (синдром Б-К родинок, синдром семейных атипичных множественных родинок-меланом). Наследование аутосомно-доминантное с высокой пенетрантностью (до 95%). Считается, что синдром диспластического невуса встречается у 2-3% здоровых людей.Множественные, разного размера (до 5-7 см) невусы располагаются как на открытых для солнечного воздействия, так и на закрытых участках тела. Часть невусов врожденная, но большинство возникает в течение жизни.
Невусы с неровными краями и неравномерной окраской, варьирующей от почти бесцветной до темно-коричневой. Гистопогически это сложные невусы. У некоторых больных невусы могут подвергаться спонтанной регрессии. У носителей синдрома описаны гормональные нарушения (например, аменорея).
Диспластический невус считается предшественником меланомы, риск возникновения которой для больных с синдромом повышен во много раз. Меланома, часто множественная, появляется у таких пациентов в более раннем возрасте, но она менее злокачественна: средняя выживаемость после удаления опухоли около 20 лет.
В связи с высоким риском возникновения меланомы больные должны находиться под наблюдением онколога. Удаление невусов без онкологических показаний вряд ли разумно, так как их число может достигать нескольких сотен. Рекомендуется фотографирование невусов с тем, чтобы можно было объективно оценить в динамике изменения в их размере и окраске и своевременно диагностировать начало малигнизации.
Синдром линейного сального невуса (синдром Ядассона) относится к факоматозам. Предполагается аутосомно-доминантный тип наследования с низкой пенетрантностью. Характерно сочетание себорейных невусов с аномалиями глаз и центральной нервной системы.
В детстве на коже лица, иногда шеи развиваются себорейные невусы. Чаще всего они локализуются в центре лба, распространяясь линейно на нос и подбородок, а также на щеки и шею. Невусы имеют вид папул желтого, светло-оранжевого и коричневого цвета. Сальные железы гипопластичны с увеличенными сосудами.
Глазные проявления включают нистагм, помутнение или утолщение роговицы, липодермоиды конъюнктивы, колобомы века, радужной и сосудистой оболочки глаза. Характерны низкий рост, диспластичные зубы и пятнистое облысение, врожденные пороки сердца и почечные гамартомы, а также атрофия коры головного мозга, умственная отсталость.
Синдром сопровождается развитием базалиом, опухолей мозга и нефробластомы. Необходимы генетическое консультирование семьи и наблюдение у детского онколога (повышен риск возникновения нефробластомы).
Альбинизм
Как правило, наследуется по аутосомно-рецессивному типу. Близкородственные браки в семьях альбиносов встречаются в 19% случаев. При альбинизме установлена генетически обусловленная неспособность меланоцитов образовывать меланин вследствие недостаточной активности тирозиназы — фермента, который в норме превращает тирозин в меланин.Фенотипические проявления синдрома весьма характерны: кожа бело-розового цвета, обесцвеченные волосы, розово-красные глаза — все это результат отсутствия пигмента в коже, луковицах волос, радужке. Отсутствие пигмента в фоторецепторах сетчатки приводит к избыточному распаду зрительного пигмента родопсина, поэтому альбиносы плохо видят днем (дневная слепота), им присуща светобоязнь.
Частичный альбинизм это отсутствие пигмента в отдельных участках, например, обесцвеченные пятна на коже, обесцвеченная прядь волос. У альбиносов часто возникают новообразования кожи, особенно на открытых участках.
С профилактической целью им рекомендуется избегать солнечного света, носить темные очки. Необходимы генетическое консультирование семьи, диспансерное наблюдение онкологом больного и облигатных и факультативных гетерозигот.
Синдром леопарда (синдром множественных лентиго)
Наследуется по аутосомно-доминантному типу, встречается редко. Характерны множественные лентиго (веснушки) и «кофейные пятна», в основном на туловище.Внешними проявлениями синдрома являются треугольное лицо, гипертелоризм, птоз, двусторонние височные шишки, крыловидная складка шеи и крыловидные лопатки, крипторхиэм и гипоспадия.
Недержание пигмента (синдром Блоха-Сульцбергера)
Наиболее вероятным считается Х-сцепленное доминантное наследование. В гомозиготном состоянии ген летален, поэтому синдром наблюдается только у женщин и проявляется при рождении или вскоре после него.На коже боковых поверхностей туловища, бедер и плеч имеются причудливой формы, резко очерченные пигментированные пятна, полосы, «брызги» с окраской от светло-коричневой до почти черной. Обычно пигментации предшествуют воспалительные явления: эритема, высыпания, пузыри.
Пигментация может существовать постоянно в течение нескольких лет или перемежаться. С возрастом она бледнеет и, как правило, исчезает к 20-30 годам. На месте пигментированных участков иногда возникает атрофия кожи.
Зубные аномалии включают широко расставленные, часто уродливые зубы, задержка прорезывания и одновременное развитие молочных и постоянных зубов. Многообразны глазные поражения: косоглазие, катаракты, пятнистая пигментация и отслойка сетчатки, атрофия глазного нерва, близорукость, голубые склеры.
Иногда встречаются гипоплазия середины лица, микроцефалия, гидроцефалия, умственная отсталость, судороги. Имеются данные об эффективности лечения большими дозами витаминов А и С в сочетании с УФ-облучением и применением кортикостероидов.
Синдром Ротмунда-Томсона
Наследуется по аутосомно-рецессивному типу, встречается редко. Синдромальная патология наблюдается рано, в 3-4 мес, редко — позже 5 лет. Первые проявления в виде нежной мраморной сетки появляются на коже лица, начиная с подбородка и распространяясь на лицо, ушные раковины, шею, конечности и ягодицы.Сетчатая пигментация постепенно становится коричнево-красной, а затем желтеет. Местами появляются атрофии, телеангиэктазии, кожные высыпания. В возрасте 3-6 лет возникают быстро прогрессирующие катаракты, почти всегда приводящие к слепоте.
К другим проявлениям синдрома относятся низкий рост, слабое оволосение, раннее поседение и облысение, ювенильный атеросклероз, дистрофические изменения зубов и ногтей, гипогонадизм (аменорея, гипоплазия яичек, крипторхизм, фистульный голос) и другие нарушения функции желез внутренней секреции. Мерой профилактики при этом синдроме является генетическое консультирование семьи. Лечение симптоматическое.
Цилиндроматоз
Цилиндроматоз (тюрбан-опухоли, кистозная железистая эпителиома Брука, трихоэпитепиома). Наиболее вероятно аутосомно-доминантное наследование с большей выраженностью у женщин, но нельзя исключить Х-сцепленное доминантное наследование.Синдром проявляется симметрично расположенными пузырчатыми эпителиомами, которые локализуются в основном на лбу, веках, щеках и на волосистой части головы. Они появляются в виде мелких бледно-желтых высыпаний, склонных к росту.
Другими проявлениями могут быть гемангиомы, одонтогенные кисты, фибромы яичников, часто отмечается гиперхолестеринемия. Синдром наблюдается преимущественно у девочек в период полового созревания. Опухоли, как правило, доброкачественные, но иногда наблюдается агрессивный рост. Профилактические мероприятия не разработаны.
Паракератозы
Наследуется по аутосомно-доминантному типу с пониженной пенетрантностью у женщин (андротропизм). Проявляется в детском или подростковом возрасте. На коже образуются центробежно распространяющиеся кератозные бляшки, окруженные узким мозолистым валиком, с атрофией в центре. Подобные же изменения наблюдаются на слизистой оболочке полости рта и гениталий. Иногда встречаются помутнение роговицы и дистрофия ногтей, часто развивается плоскоклеточный рак кожи.Из более редких форм наследственного или предполагаемого наследственного рака кожи можно назвать следующие.
Пузырчатка Брокка
Пузырчатка Брокка (буллезный дистрофический эпидермолиз) образуются пузыри на коже и слизистых оболочках с последующей мокнущей эрозией и развитием рубцов, подверженных перерождению в плоскоклеточный рак кожи. Патогенез неясен.Синдром Ван-ден-Боша
Синдром Ван-ден-Боша — проявляется дистрофией кожи с ангидрозом, алопецией, задержкой роста, снижением интеллекта и кератозом кистей и стоп с бородавчатыми разрастаниями, склонными к малигнизации. Наследование рецессивное, Х-сцеппенное.Болезнь Дарье-Уайта
Болезнь Дарье-Уайта (фолликулярный дискератоз). При этом заболевании нарушается процесс ороговения кожи, развиваются конические папулы, закономерно перерождающиеся в кожные карциномы. Наследование аутосомно-доминантное.Врожденный дискератоз
Врожденный дискератоз (синдром Цинссера-Энгмана-Коула) проявляется пойкилодермией с диссеминированным кератозом, лейкоплакией слизистой полости рта, разрушением ногтей, атрофической эритемой кожи ладоней и подошв, закупоркой сальных желез.Для этого заболевания характерны плоскоклеточные карциномы кожи. Описаны две формы наследования: аутосомно-доминантная и Х-сцеппенная рецессивная.
Порфирия наследственная
Связана с нарушением биосинтеза порфиринов в печени и костном мозге. Отмечена повышенная светочувствительность кожи с гиперпигментацией и гипертрихозом. У больных нередко развивается плоскоклеточный рак кожи. Наследование аутосомно-доминантное.Пахионихия врожденная
Характеризуется деформацией, утолщением и потемнением ногтей, кератозом ладоней и подошв, кожи локтей и коленей, языка, гипергидрозом, умственной отсталостью. Может сопровождаться раком слизистой полости рта, а у женщин также наружных половых органов. Наследование аутосомно-доминантное.Предраковый меланоз Дюбрейля-Хатчинсона
Возникает в пожилом возрасте в виде небольшого пигментного пятна на коже лица, груди или кисти, или слизистой полости рта. Пятно неравномерно окрашено, инфильтрировано, в 40% случаев развивается меланома. Характер наследования не изучен. Болеют чаще женщины.Синдром нейрокожного меланоза иногда выделяют из множественной эндокринной неоплазии (МЭН) 2 типа. Для него помимо эндокринных аденом характерны множественные, в некоторых случаях гигантские пигментные пятна, склонные к перерождению в злокачественную меланому.
Угляница К.Н., Луд Н.Г., Угляница Н.К.
Опубликовал Константин Моканов