Синдром луи бар что это такое
Синдром Луи-Бар - причины, симптомы, диагностика и лечение
Синдром Луи-Бар (атаксия-телеангиэктазия) — наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Общие сведения
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
Синдром Луи-Бар
Причины и патогенез синдрома Луи-Бар
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Клинические проявления синдрома Луи-Бар
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
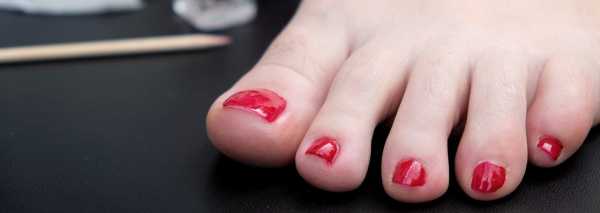
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
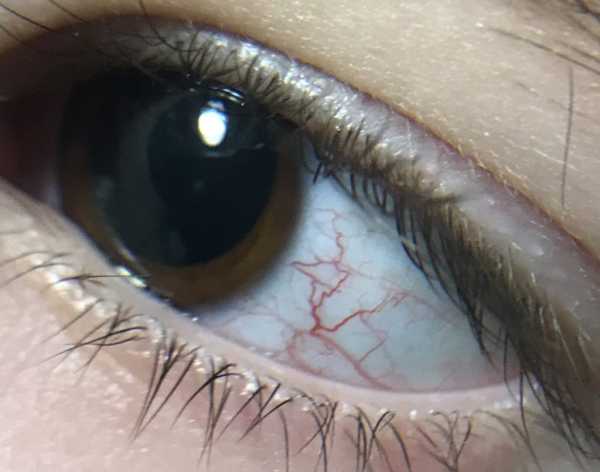
Синдром Луи-Бар характеризуется изначальным возникновением телеангиэктазий на конъюнктиве глазного яблока, где они имеют вид «паучков». Затем сосудистые звездочки появляются на коже век, носа, лица и шеи, локтевых и коленных сгибов, предплечий, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба. Наиболее выражены сосудистые звездочки в тех местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне или проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома. Особенностью онкопатологии в случае синдрома Луи-Бар является повышенная чувствительность пациентов к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при их лечении.
Диагностика синдрома Луи-Бар
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться: УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю-Ослера, атаксией Пьера-Мари, болезнью Гиппеля-Линдау и др.
Лечение и прогноз синдрома Луи-Бар
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
В связи с отсутствием эффективных способов лечения синдром Луи-Бар имеет неблагоприятный прогноз как для выздоровления, так и для жизни. Больные этим заболеванием редко доживают до 20 лет. В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
проявления, выбор диагностики и симптоматическое лечение
Синдром Луи-Бар, известный также как атаксия-телеангиэктазия, – врожденная патология, имеющая генетическую природу. Нарушения формируются еще на раннем этапе развития плода и связаны с дефектом строения хромосомы. Клинические проявления заболевания в большинстве случаев специфические и позволяют поставить диагноз в короткие сроки. Дети с синдромом Луи-Бар страдают от двигательных расстройств на фоне дефектов строения мозжечка, у них диагностируется сосудистый рисунок на коже, слизистых и склере глаз. Поражается и иммунная система, что проявляется частыми инфекционными и вирусными заболеваниями. Лечение патологии на сегодняшний день не разработано, терапия носит симптоматический характер. В связи с этим прогноз при наличии недуга неблагоприятный.
Причины развития синдрома Луи-Бар
Основу заболевания представляет генетическая мутация, что обеспечивает формирование отклонений еще в первом триместре беременности. Происходит изменение строения плеча 11 хромосомы. Именно этот дефект и провоцирует развитие клинических признаков синдрома Луи-Бар у детей. При этом патология формируется в тех случаях, когда оба родителя являются носителями мутации. Это связано с тем, что заболевание имеет аутосомно-рецессивный тип наследования. Точные причины, провоцирующие развитие нарушения, на сегодняшний день неизвестны. Предположительно, спровоцировать у ребенка развитие синдрома Луи-Барра может пагубное воздействие на хромосомный набор, вызванное стрессом у матери на ранних сроках беременности, а также воздействие ионизирующего излучения.

Основные проявления недуга
Главными мишенями генетической аномалии являются структуры головного мозга и иммунная система человека. Именно с их поражением связано большинство клинических признаков заболевания. Синдром Луи Барра имеет несколько основных симптомов, которые считаются патогномоничными, то есть позволяют поставить диагноз. В ряде случаев у младенцев и детей школьного возраста отмечаются и другие проявления патологии, встречающиеся не так часто.
Мозжечковая атаксия
В результате генетической мутации нарушается процесс закладки нервной трубки. Это сопровождается дефектами различных отделов головного мозга. Наиболее выраженным изменениям подвержены мозжечок, некоторые участки коры и черная субстанция. Подобные нарушения сопровождаются специфической симптоматикой. Она проявляется у ребенка в возрасте от 5 месяцев до 3–4 лет. Данная особенность связана с тем, что именно в этот период малыши начинают активно ползать и учиться ходить. У пациентов отмечается выраженная атаксия, то есть неустойчивость вплоть до полной невозможности удерживать равновесие. В ряде случаев синдром Луи-Бар сопровождается и нарушением речи, которая представляется невнятной. Данный дефект также обусловлен аномалиями развития мозжечка. В связи с такими изменениями наблюдается мышечная слабость, снижение сухожильных рефлексов.
Телеангиэктазия
Термин обозначает расширение поверхностных мелких капилляров и венул кожи, склеры и слизистых оболочек, что сопровождается формированием специфичных «узоров» и сосудистой сеточки. Данный симптом проявляется у детей, как правило, в возрасте с 3 до 6 лет, в редких случаях возникает позднее. Такое клиническое проявление свойственно и многим других заболеваний. Однако в сочетании с атаксией этот признак позволяет подтвердить наличие синдрома Луи-Бара.
Телеангиэктазия наблюдается преимущественно на лице, склере глаз, а также в области локтевых и коленных сгибов. Интенсивность проявления сосудистых звездочек повышается при воздействии солнечных лучей. Зачастую данный дефект сочетается с сухостью кожи, гипертрихозом и изменениями, по внешнему виду напоминающими псориаз.
Проблемы с иммунитетом и дыханием
Защитные силы организма на фоне синдрома Луи-Бар значительно ослабевают. Это происходит за счет снижения производства иммуноглобулинов и Т-лимфоцитов. Данные соединения играют важную роль в поддержании клеточного иммунитета.
На фоне снижения защитных сил организма отмечается частое развитие инфекционных процессов, преимущественно поражающих респираторную систему. Дети страдают от ринитов, синуситов, пневмонии и других заболеваний. Для таких патологий характерно длительное течение, а также устойчивость к антибактериальной терапии.
Новообразования
Отдельное место синдрому Луи-Бар в иммунологии отводится еще и потому, что генетическое расстройство зачастую сопровождается высоким риском развития опухолей. Данные процессы чаще всего диагностируются в лимфоретикулярной системе. У пациентов регистрируются раковые поражения красного костного мозга, с трудом поддающиеся лечению. Усугубляется положение и тем фактом, что детям с синдромом Луи-Бар противопоказано использование лучевой терапии. Распространенным заболеванием при данной патологии является лимфома.
Зрение
Телеангиэктазия отмечается не только на коже, но и в мембране, покрывающей склеру глаза. Этот симптом сочетается с поражениями связочного аппарата данного анализатора. Нарушается процесс координации кривизны хрусталика. Вследствие дефектов у детей развивается косоглазие, может снижаться острота зрения.
Ортопедические отклонения
У большинства малышей с атаксией-телеангиэктазией отмечается деформация стоп, что лишь усугубляет двигательные расстройства, поскольку пациентам тяжело переносить вес тела с одной конечности на другую. В ряде случаев диагностируются и различные искривления позвоночника, при этом ярко выраженные проблемы встречаются редко. В случае синдрома Луи-Бара эти дефекты хорошо поддаются хирургической коррекции.
Диагностика
Подтверждение наличия заболевания начинается с осмотра пациента и сбора анамнеза. Патогномоничным считается сочетание нарушений координации с телеангиэктазией. При этом основу диагностики генетических проблем составляет анализ ДНК пациента, который позволяет выявить аномалию строения хромосомы. С точки зрения иммунологии важным считается осуществление анализов крови, в которых выявляют ряд характерных изменений. Они включают в себя:
- Снижение количества лимфоцитов. Это происходит преимущественно за счет уменьшения выработки Т-клеток.
- Недостаточная концентрация иммуноглобулинов. При синдроме Луи-Бар чаще отмечается низкое содержание фракций IgA и IgE.
- Поскольку у некоторых пациентов заболевание сопровождается также симптоматикой аутоиммунных нарушений, отмечается наличие в крови соответствующих комплексов: аутоантител к иммуноглобулинам и митохондриям.
Визуальные методы, позволяющие сделать фото внутренних органов, также широко применяются. Используется УЗИ, МРТ и рентген. Для комплексной оценки состояния больного потребуется консультация врачей различной специализации, начиная с иммунолога и заканчивая ортопедом.

Лечение и прогноз
Специфической терапии, которая позволила бы побороть синдром Луи-Барра, нет. Поэтому борьба с заболеванием носит симптоматический характер. Лечение направлено в первую очередь на недопущение развития инфекционных поражений, которые становятся распространенной причиной гибели пациентов. К летальному исходу приводят и онкологические процессы, контролировать которые очень сложно. Для коррекции состояния пациентов применяются антибиотики, кортикостероидные препараты, витамины и внутривенные инфузии.
Современные концепции лечения недуга основаны на следующих принципах:
- Для борьбы с неврологическими расстройствами используются препараты леводопы, антагонисты дофамина и антихолинергические средства. Коррекция тремора производится медикаментами типа «Габапентина», а «Флуоксетин» и «Буспирон» используются с целью уменьшения интенсивности речевых нарушений.
- Во многих случаях оправдано назначение парентерального питания. Это особенно актуально у маленьких пациентов в период лечения инфекционных поражений.
- Для предотвращения развития септических процессов и осложнений со стороны респираторной системы используются антибиотики широкого спектра действия. Ряд врачей склоняется к оправданности их профилактического назначения.
- Рентгенологические исследования у пациентов с генетическим дефектом строго ограничены. По возможности рекомендуется применять альтернативные методы, например, магнитно-резонансную томографию или ультразвук.
- Для контроля онкологических процессов в организме больных требуется регулярный скрининг. Он подразумевает как проведение стандартных анализов крови, так и тестов с использованием специфических маркеров, которые позволяют распознать опухолевый очаг в лимфоретикулярной системе.
Прогноз при синдроме Луи-Бар неблагоприятный. Большинство больных погибают в 20–25 лет. При этом в 65–70% случаев причиной смерти становится хроническое поражение легких. Инфекции склонны к переходу в септический процесс.
Отзывы
Марина, 32 года, г. Ростов-на-Дону
Сын родился с синдромом Луи-Бар. Это стало заметно, когда он начал учиться ходить. Все время падал, не держал равновесие. Позднее появилась сосудистая сеточка на лице. Врачи поставили предварительный диагноз уже после осмотра. Он подтвердился результатами анализов. Прогноз при болезни неблагоприятный. Сейчас принимаем витаминно-минеральный комплекс, надеемся на лучшее.
Федор, 29 лет, г. Петропавловск-Камчатский
Долго не могли научить дочку ходить. Делала шаг и падала, дрожала голова. Решили обратиться за помощью к врачу. Сдали анализы крови, сделали УЗИ. Доктор поставил диагноз «синдром Луи-Бар». Это генетическая проблема, лечения которой нет. Будем делать все возможное, чтобы поддерживать здоровье дочки.
Загрузка...Синдром Луи-Бар: симптомы, диагностика и лечение
Как известно, существует много различных хромосомных аномалий, которые закладываются еще в период внутриутробного развития. Изучением этих патологий занимаются генетики. В последние годы данная область медицины активно развивается, поэтому в скором будущем такие заболевания легче будет диагностировать и лечить. К счастью, данные аномалии встречаются очень редко. Это связано с улучшением диагностики плода. Одной из патологий, связанных с хромосомными нарушениями, является синдром Луи-Бар. В большинстве случаев это заболевание выявляют в первый год жизни младенца, но иногда оно дает о себе знать лишь к 6-7 годам.

Синдром Луи-Бар – что это за патология?
Эта патология относится к врожденным генетическим порокам. В большинстве случаев она передается по наследству. Атаксия-телеангиэктазия (синдром Луи-Бар) встречается крайне редко. Данное заболевание имеет специфичные проявления, которые позволяют диагностировать эту патологию. Чтобы поставить точный диагноз, необходим консилиум врачей, которые подтвердят или опровергнут наличие страшной аномалии.
История и эпидемиология заболевания
Данный синдром встречается очень редко. Его частота составляет около 1 случая на 40 тысяч населения. Впервые заболевание было обнаружено французской женщиной-ученым Луи-Бар. Синдромы, характерные для данной патологии, она объединила в одну нозологию. Это произошло в 1941 году. После было обнаружено еще несколько случаев заболевания по всему миру. Так как данная аномалия встречается крайне редко, нельзя с точностью сказать, какова этиология синдрома Луи-Бар. Считается, что появление заболевания не зависит от климатических условий. Поэтому синдром может встречаться в любых регионах. Помимо этого, нет данных, которые связывали бы заболеваемость с полом пациента. То есть синдром Луи-Бар с одинаковой частотой наблюдается как у мальчиков, так и у девочек.
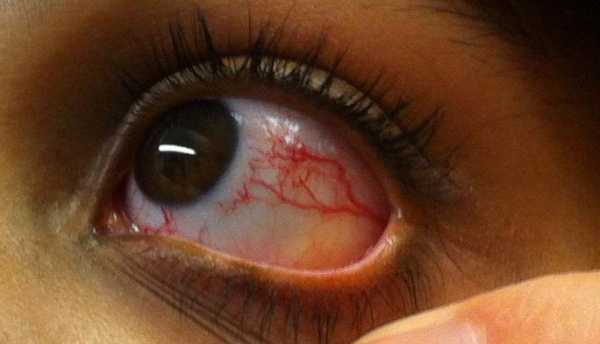
Причины развития патологии
Данная аномалия развития закладывается еще в первом триместре беременности. Заболевание передается только по наследству. Синдром относится к аутосомно-рецессивным генетическим патологиям. Это означает, что ребенок точно унаследует заболевание, если оба родителя имеют нарушение хромосом. Если же аномалия наблюдается у одного из них (независимо от пола), то шанс появления синдрома Луи-Бар у малыша составляет 50%. Основная причина мутации – это нарушение длинного плеча 11-й хромосомы. Точные факторы, которые приводят к такой генетической перестройке, неизвестны. Но выделяют ряд вредных воздействий, влияющих на эмбриональное развитие. В первую очередь это факторы окружающей среды (облучение, отравление ядовитыми веществами). Также в первом триместре беременности очень опасен стресс.

Синдром Луи-Бар: патогенез заболевания
Как и большинство врожденных хромосомных патологий, данный синдром охватывает сразу несколько органов и систем. Основными мишенями этого заболевания являются головной мозг и иммунитет человека. Также имеется и выраженное поражение кожного покрова. Все клинические проявления данного заболевания связаны с механизмом его развития. В первую очередь наблюдаются дегенеративные процессы в ЦНС. А именно, мозжечковая атаксия. При этом часть элементов не развивается (волокна Пуркинье и зернистые клетки). Другими видимыми нарушениями являются кожные проявления – телеангиэктазии. Они представляют собой расширенные сосуды, которые особенно выражены на лице (инъекция склер, ушные раковины, нос). Атаксия мозжечка и телеангиэктазии в совокупности носят название синдром Луи-Бар. Детей, рожденных с данным заболеванием, можно выделить в первые годы жизни, так как аномалия проявляется выраженными физическими нарушениями (отставание в развитии, неустойчивое положение тела, слабость мышц).

Помимо этого, патогенез заболевания включает в себя недостаточность иммунной системы (Т-лимфоцитов). У детей, страдающих данной патологией, наблюдается гипо- или полная аплазия тимуса. В результате этого клеточный иммунитет развит очень слабо и не способен обеспечивать защиту организма от инфекционных процессов.
Симптомы атаксии-телеангиэктазии
Выраженность клинической картины зависит от степени поражения мозжечка и гипоплазии вилочкой железы. Это и определяет, как будет проявляться синдром Луи-Бар. Симптомы заболевания:
- Мозжечковая атаксия. Данный синдром проявляется раньше других, обычно на первом году жизни. Он становится выражен к моменту начала самостоятельной ходьбы. Дети с атаксией мозжечка зачастую не могут стоять и нормально передвигаться. В более благоприятных случаях наблюдается шаткость походки и тремор конечностей. Помимо этого, неврологическая симптоматика выражается в слабости мышц, дизартрии различной степени (невнятная речь) и косоглазии.
- Телеангиэктазии. Кожные проявления синдрома Луи-Бар менее опасны. Обычно они дают о себе знать в возрасте от 3 до 6 лет. Телеангиэктазии – это расширенные капилляры, которые носят название «сосудистые звездочки». Больше всего они заметны на открытых участках тела, в частности на лице. Расширенные сосуды часто находятся в глазах, на носу и ушах, а также сгибательных поверхностях рук и ног.
- Склонность к инфекциям. Из-за выраженного иммунодефицита организм не справляется с вредными агентами самостоятельно. В результате у ребенка часто развиваются различные инфекции. Зачастую это хронические заболевания дыхательных путей – фарингит, ларингит, тонзиллит, пневмония.
- Опухолевые процессы. Из-за гипоплазии тимуса, помимо инфекционных процессов, организм становится восприимчив к раковым заболеваниям. Чаще всего это опухоли кроветворной и лимфоидной ткани. Если синдром Луи-Бар у ребенка является достоверным диагнозом, то ему строго запрещено лечение рака ионизирующим облучением.
Диагностика атаксии-телеангиэктазии
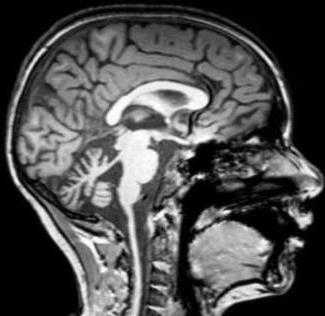
Диагностика синдрома Луи-Бара обычно не предоставляет большой сложности, так как его симптомы довольно специфичны. Заподозрить данное заболевание можно с первых лет жизни по клинической картине. Неврологическая симптоматика (мозжечковая атаксия, мышечная слабость, тремор и косоглазие) в совокупности с телеангиэктазиями являются показанием для диагностики этой патологии.
При подозрении на синдром Луи-Бар необходима консультация сразу нескольких специалистов. Среди них: невропатолог, дерматолог, онколог, инфекционист, эндокринолог и генетик. Помимо клинического обследования, выполняется лабораторная и инструментальная диагностика. Проводят иммунологические анализы, в которых отмечается уменьшение или полное отсутствие элементов клеточного иммунитета (снижение Т-лимфоцитов, иммуноглобулинов А, G). В ОАК наблюдается лейкоцитоз и ускорение СОЭ, что говорит о воспалительном процессе в организме. Также важна и инструментальная диагностика. Выполняется рентгенография грудной клетки (уменьшение размеров тимуса), МРТ головного мозга (дегенеративные процессы). В настоящее время, помимо стандартных исследований, проводят генетическое (исследуют нарушение 11-й хромосомы), на основании которого ставится точный диагноз.
Лечение синдрома Луи-Бар
К сожалению, этиологического лечения хромосомных аномалий на данный момент не разработано. Поэтому при данной патологии проводят лишь симптоматическую терапию и постоянное наблюдение за больным. В первую очередь лечение направлено на улучшение работы иммунной системы. Это необходимо, чтобы избежать инфекций и опухолевых процессов. С данной целью используют гамма-глобулин и препарат «Т-активин». При развитии воспалительных заболеваний применяют антибактериальные и противовирусные средства. К сожалению, синдром мозжечковой атаксии не поддается полному лечению. Чтобы приостановить дегенеративные процессы, используют ноотропные препараты. При онкологических заболеваниях прибегают к химиотерапии и хирургическому лечению.
Прогноз для жизни при синдроме Луи-Бар
Несмотря на тяжесть заболевания, при своевременной диагностике и лечении можно продлить и облегчить жизнь ребенка. С этой целью разработана паллиативная терапия для таких пациентов. К сожалению, аномалия Луи-Бар может прогрессировать быстро. В этом случае продолжительность жизни составляет 2-3 года. Иногда заболевание не развивается в течение нескольких лет. При этом продолжительность жизни значительно увеличивается. Максимальным возрастом пациентов считается 20-30 лет. В большинстве случаев причинами смерти являются инфекционные и опухолевые процессы, иногда – неврологические нарушения.
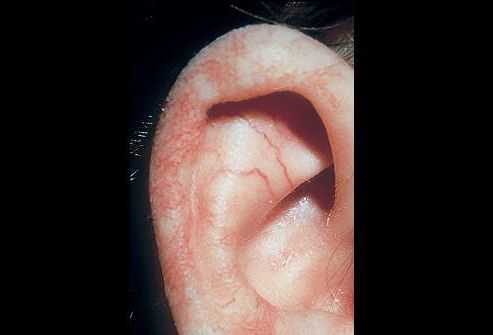
Профилактика синдрома Луи-Бар
Чтобы избежать развития данной патологии, необходимо проводить генетическое обследование плода еще на ранних сроках беременности. Также важно знать анамнез не только родителей будущего ребенка, но и других членов семьи. Во время беременности нужно избегать вредного воздействия окружающей среды и психоэмоциональных стрессов.
Если малыш с такой аномалией уже родился, то важно выполнять все назначения врача, защитить ребенка от инфекционных агентов. При слабом иммунитете и нарушенном физическом развитии необходимо своевременно диагностировать синдром Луи-Бар. Фото детей с данным заболеванием можно увидеть в специальной медицинской литературе.
Синдром Луи-Бар: симптомы, признаки, диагностика и лечение
Описание
Синдром Луи Бар в медицинской практике встречается не так часто, но, тем не менее, современные медики особенно опасаются данного заболевания. Это наследственная болезнь, связанная с иммунодефицитом, которая распространяется исключительно по аутосомно-рецессивному типу. В ходе патологического процесса преобладает одно из двух поражений иммунной системы, в частности страдает клеточный иммунитет. Такие потери в организме невосполнимы, а обеспечить пациенту полноценную жизнь порой просто нереально.

Рассуждая о патогенезе синдрома Луи Бар, стоит отметить, что пациентам с таким диагнозом свойственно отсутствие тимуса, а также недоразвитость лимфатических узлов и селезенки. Кроме того, до конца не сформированы органы переферии иммунной системы, вызывающие, тем самым, патогенное воздействие на человеческий ресурс со стороны различных микроорганизмов.
Причина данной патологии очевидна – генетический дисбаланс, на фоне которого еще во внутриутробном периоде преобладает нейроэктодермальная дисплазия. Имея аутосомно-рецессивное происхождение, характерный недуг передается в случае получения рецессивного гена от обоих родителей сразу.
На фоне такой аномалии прогрессируют дегенеративные изменения мозжечка, которые непосредственно затрагивают его зубчатое ядро, черную субстанцию и определенные "звенья" коры головного мозга. Такой обширный радиус действия просто не может не отразиться на генетическом и молекулярном уровне, а новорожденный появляется на свет со страшным диагнозом.
В этиологии синдрома Луи Бара также преобладает врожденный дефицит IgA и IgE, что влечет за собой учащение инфицирования организма и продолжительное лечение преобладающих заболеваний. Нарушенный на генетическом уровне иммунитет также чреват формированием злокачественных опухолей и раковых клеток. Так что крайне важно подробная диагностика и своевременное лечение маленького пациента.
Симптомы
Как правило, симптомы синдрома Луи Бара начинают проявляться в возрасте пяти месяцев – трех лет, но особенно заметны отклонения, когда малыш начинает самостоятельно передвигаться пусть и не на дальние расстояния.
Так, признаки атаксии на лицо: шаткая и неуверенная походка, нарушенная координация движений, тремор конечностей, качание туловища и частое подергивание головы. Характерные признаки в пораженном организме зачастую настолько очевидны, что пациент просто не способен самостоятельно передвигаться. Кроме того, имеет место нарушенная речь, отсутствие сухожильных рефлексов, мышечная гипотония, косоглазие и прочие отклонения в структуре и функциональности глаз.
При данном заболевании очень часто прогрессируют инфекционные заболевания дыхательных путей и уха рецидивирующего характера. Это может быть ринит хронической формы, отит, фарингит, синусит, бронхит, реже – воспаление легких и пневмония. Однако важно понимать, что каждый последующий рецидив лишь ухудшает общее состояние, приближая летальный исход.
Еще одним красноречивым симптомом синдрома Луи Бара являются сосудистые звездочки, которые появляются, как правило, в 3 - 6 летнем возрасте. Они спровоцированы патогенным расширением небольших капилляров, однако могут свидетельствовать и о наличии других заболеваний.
Начинается телеангиэктазия на глазном яблоке виде тривиального конъюнктивита, однако уже очень скоро характерный визуальный дефект преобладает на коже век, шеи, носа, лица, локтях и тыльной стороны кисти. Также преобладает повышенная сухость кожных покровов, гиперемия, раннее выпадение волос и увеличение числа сосудистых сеточек на кожных покровах.
Синдром Луи Бара может сопровождаться появлением злокачественных новообразований, представленных лимфомой и лейкемией. Однако клинику данных патологических процессов желательно изучать в индивидуальном порядке.
Диагностика
Если у участкового терапевта возникло подозрение на присутствие синдрома Луи Бара, то он направляет его к узкому специалисту. Однако консультации у иммунолога вовсе недостаточно, ведь также стоит показаться со своей проблемой неврологу, дерматологу, офтальмологу, пульмонологу, онкологу и отоларингологу. СКрайне важно дефференцировать синдром Луи-Бара с болезнью Рандю-Ослера, атакой Фридрейха, атаксией Пьера-Мари и, конечно, мало изученным синдромом Гиппеля-Линдау.

Ставить окончательный диагноз будет невролог, однако без подробной диагностики сделать это нереально. Именно поэтому обязательно необходимо пройти инструментальное и лабораторное исследование для получения развернутой клинической картины.
Самые востребованные методы обследования представлены ниже:
- в общем анализе крови можно наблюдать патологическое снижение количества лимфоцитов;
- определение уровня иммуноглобулинов крови позволяет выявить снижение IgA и IgЕ, а также достоверно определить присутствие аутоантител к митохондриям, иммуноглобулину и тиреоглобулину;
- УЗИ помогает охарактеризовать аплазию и гипоплазию тимуса;
- МРТ головного мозга диагностировать деградацию мозжечка и патогенное расширение IV желудочка;
- Рентгенография определяет присутствие пневмонии, очагов пневмосклероза, а также преобладание бронхоэктатических изменений.
Когда все результаты диагностика, а также предварительное заключение узких специалистов будут у невролога на руках, он наконец-то определиться с окончательным диагнозом и назначит определенную схему лечения.
Профилактика
Профилактические меры не отличаются особой эффективностью, поскольку патологической процесс преобладает при непосредственном формировании эмбриона во внутриутробном периоде.
Болезнь передается по наследству и преобладает на генетическом уровне, поэтому оградить своего будущего ребенка от страшного рока весьма проблематично.
Врачи при выявлении характерной проблемы на одном из скринингов во время беременности, предлагают будущей мамочке преждевременно стимулировать роды.
Лечение
В современной медицине так и не обнаружена панацея от данного заболевания, да что там говорить, медики не могут даже определиться с общей схемой лечения. Однако в данной клинической картине однозначно требуется комплексный подход.
- Необходим продолжительный курс антибактериальной терапии, который позволяет в кратчайшие сроки истребить вторичные бактериальные инфекции, как основную причину иммунодефицита.
- Наряду с приемом антибиотиков требуется и курс гамма-глобулинов, иммуностиммуляторов, поливитаминных комплексов и даже БАДов для всеобщего укрепления ослабленного человеческого ресурса.
- В детском возрасте обязательна физиотерапия, представленная индивидуальными занятиями с логопедом по постановке речи.
Однако, так или иначе, терапия должна базироваться на основном заболевании. Если это сахарный диабет, то в схеме лечения не обойтись без пероральных сахароснижающих препаратов и инсулина. Если имеет место стремительно прогрессирующая опухоль, то требуется незамедлительное ее удаление хирургическим путем. Так что при лечении важно учитывать все нюансы, и тогда он будет, действительно, эффективным.
Луи-Бар синдром
Луи-Бар синдром (синонимы: атаксия-телеангиэктазия, цефало-окуолокутанная телеангиэктазия.)
В 1941 г. Луи-Бар описала синдром, характеризующийся мозжечковой атаксией и кожно-конъюнктивальной телеангиэктазией. В 1958 г. Boder и Sedgwick предложили название атаксия-телеангиэктазия и добавили третий важный компонент этого синдрома - рецидивирующую тяжелую синопульмональную инфекцию.
Этиология и патогенез
Этот синдром в различных классификациях рассматривается как спинно-мозжечковая дегенерация (Toller и Millichap) или как факоматоз (Boder, Miller и др.). Термин факоматоз (phakomatosrls) предложил van der Hoeve в 1923 г. для заболеваний с комбинированным поражением нервной системы и кожи (врожденные нейро-экто-мезодермальные дисплазии) Boder и Sedgwick в 150 случаях синдрома Луи-Бар обнаружили дегенеративные явления в ЦНС, дегенерацию пирамидальных клеток, расширение менингеальных сосудов и др.
Значительная часть описанных наблюдений относится к семейно-наследственной патологии, тип наследования аутосомно-рецессивный.
Многие современные дерматологи синдром Луи-Бар относят к сочетанным иммунодефицитным состояниям. В основе лежат гипоплазия тимуса и дефицит IgA и IgE, т. е. нарушение функции клеточного и гуморального звеньев иммунитета, чем и объясняют частые рецидивирующие инфекционные заболевания органов дыхания, пищеварительного тракта, кожи и др. Помимо гипоплазии вилочковой железы отмечается гипо- атрофия лимфатических узлов, селезенки и лимфатического аппарата, пищеварительного канала. Снижение иммунитета приводит к ранней гибели больных от инфекционных заболеваний и усиливает тенденцию к развитию злокачественных новообразований лимфатической системы.
Клиника
Заболевание встречается редко. Оно включает в себя поражения нервной системы, кожно-конъюнктивальные телеангиэктазии и инфекционные заболевания. Среди неврологических манифестаций этого синдрома атаксия мозжечкового характера является первым симптомом у всех больных; начинается с раннего детства, становится особенно заметной, когда ребенок начинает ходить. Вследствие прогрессирования мозжечковых расстройств больные часто полностью лишены возможности ходить. Отмечается атаксия позы и конечностей, гипотония мышц, замедленная скандированная невнятная речь по типу мозжечковой дизартрии, нистагм, тремор, нарушения иннервации движений глазных яблок. Сухожильные рефлексы низкие или отсутствуют. Окуломоторная апрексия встречается в 80% случаев, затем развивается офтальмоплегия.
Телеангиэктазии обычно появляются после атаксии, в возрасте 4-6 лет, в некоторых случаях позже, а иногда на первом месяце жизни. Телеангиэктазии вначале развиваются на глазных яблоках (бульбарная конъюнктива), затем на веках и коже лица.
Инфекционные заболевания главным образом органов дыхания обнаруживаются у 80% больных. Они проявляются рецидивирующими рино-фарингитами, хроническими бронхитами, бронхопневмониями, бронхоэктазиями. Описываются гнойные отиты. Патогенетически их связывают с иммунным дефицитом.
Дерматологическая картина синдрома Луи-Бар характеризуется наличием кожных телеангиэктазий у 100% больных. Другие кожные проявления (резкая сухость кожи и волос, фолликулярный кератоз на предплечьях и голенях, пигментации цвета «кофе с молоком» и сетчатые дисхроматически-атрофические поражения на коже лица) наблюдаются у 50-70% больных.
Кожные изменения не являются специфическими для атаксии - телеангиэктазии, однако диагностическая важность этих изменений делает необходимым их раннее выявление в целях более успешного лечения; часто диагноз ставится на основании дерматологической картины.
Вначале телеангиэктазии появляются на бульбарной конъюнктиве, затем распространяются на кожу век, носа, ушных раковин, шеи; появляются на коже локтевых сгибов, предплечий, подколенных областей, тыльной поверхности кистей, стоп. Телеангиэктазии особенно выражены наг участках кожи, которые подвергают
Синдром луи бар (атаксия телеангиэктазия): причины и лечение патологии
Впервые синдром Луи-Бара был замечен и описан на территории Франции в 1941 году. С тех пор его частота появление заметно возросла и начала встречаться по всему земному шару.
Статистика говорит, в современном обществе шанс иметь этот синдром имеет 1 человек из 40 тысяч населения.
Его суть заключается во врожденном неправильном иммунном состоянии организма, который в частности поражает Т-звено и начинает проявляться в аномальных изменениях во всем организме.
Люди, страдающие синдромом, склонны к частым инфекционным заболеваниям, а также имеют высокие шансы на развитие злокачественных опухолей по всему организму.
Чаще всего если синдром Луи-Бар начинает проявляться у детей при самом рождении, то это чревато смертельным исходом даже без шансов правильно и вовремя диагностировать такого больного.
Болезнь в одинаковой пропорции поражает как мужчин, так и женщин, максимально быстро разрушая их нервную систему и кожные покровы.
Причины возникновения
Возникать синдром может на генетическом уровне, при малейших сбоях или отклонениях от нормы.
Подобный сбой чреват нейроэктодермальной дисплазией, которая является врожденной у таких людей.
Патологию относят к аутосомно-рецессивным заболеваниям, которое может проявится, если генные нарушения присутствовали одновременно у обоих родителей.
Заболевание склонно полностью менять и разрушать ткани мозжечка, добираясь даже до его ядра.
Подобные ситуации приводят к дегенеративным изменениям в коре головного мозга, а также спинномозговых путей.
Синдром Луи-Бара часто часто сочетается с другими генетическими заболеваниями и тщательно за ними скрывает свои признаки.
Проявить его удается только после длительных и затруднительных лечений инфекционных болезней, которые не дают желаемого результата.
Сильные иммунные нарушения приводят к образованию злокачественных опухолей, которые берут свое начало в лимфоретикулярной системе.
Симптоматика синдрома
В современной медицине патология встречается довольно редко, однако врачи опасаются возможного развития 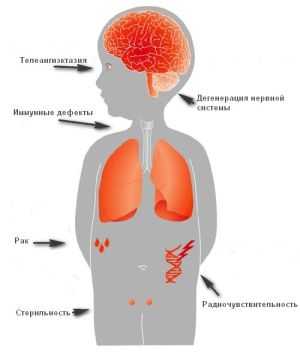 заболевания.
заболевания.
Так как эта генетическая болезнь частично или полностью разрушает клеточный иммунитет, имеет патологический характер и не поддается лечению. Полноценная жизнь практически нереальна.
Симптоматика заболевания во взрослом возрасте может выражаться не сразу.
Чаще всего ее выявляют по постепенному ухудшению работы внутренних органов, поражении иммунной системы, полном или частичном отсутствии тимуса.
Если синдром Луи-Бара развивался внутриутробно, поражая мозжечок и кору головного мозга ребенка, то новорождённый с самого появления на свет имеет дегенеративные изменения и обрекающий на мучения диагноз.
Если при рождении у малыша не были заметны первые признаки болезни, то уже в возрасте 3-24 месяцев синдром начнет проявляться достаточно ускоренно.
Чаще всего это выражается в полном отсутствии движений, плохой координацией, застоем умственного развития и внешними признаками развития лица и конечностей.
Это может быть:
- мышечная гипотония;
- косоглазие;
- отсутствие рефлексов и функциональности мышц и глаз.
Синдром Луи-Бара часто проявляется в постоянных инфекционных заболеваниях, которые касаются дыхательных путей и ушей.
Это может быть отит, фарингит, бронхит, синусит и другие заболевания.
Пневмония и воспаление легких при этом практически никогда не проявляются. Каждое последующее заболевание имеет более острую форму и осложнения, которые не поддаются лечению.
Признаком симптома принято считать и сосудистые звездочки, которые могут появляться по всему телу в возрасте от 3 лет.
Чаще всего это связанно с расширением капилляров, однако при наличии только этого симптома нужно искать и другие варианты возможных заболеваний.
Что касается внешнего облика лица и глаз, то здесь в первую очередь начинает проявляться телеангиэктазия на глазном яблоке.
Чревато это постоянными конъюнктивитами, визуальные признаки которого могут проявляться не только на глазах, но и на шее, щеках, ушах, веках и даже на ладонях.
Кроме этого кода всего тела становится сухой и шелушится, обильно выпадают волосяные покровы.
В самых запущенных ситуациях синдром может спровоцировать злокачественные образования, лейкемию и лимфому.
Что делают для диагностики?
При первых признаках или подозрениях на болезнь этого рода, любой врач делает назначение и направление на доктора более узкой специализации.
Довольно часто таких больных одновременно наблюдает несколько врачей, которые назначают лечение совместным консилиумом.
Это может быть иммунолог, дерматолог, офтальмолог, невролог, онколог и отоларинголог. Только их совместные консультации смогут отличить этот симптом от других редких и опасных видов заболеваний.
Окончательный диагноз при таком заболевании всегда ставит только невролог, если у него на руках будут все результаты клинических анализов и лабораторных исследований.
Чаще всего установить диагноз помогают определенные показатели, которые не соответствуют норме. В частности, в крови могут полностью отсутствовать лимфоциты, а уровень иммуноглобулина будет намного ниже нормы.
При этом совершенно будут отсутствовать любые антитела для борьбы с вирусными инфекциями и заболеваниями.
Кроме этого о наличии синдрома может сказать снимок УЗИ, МРТ и рентгенография, где будут видны размеры и само наличие тимуса, мозжечка и очагов злокачественных опухолей.
Когда окончательный диагноз будет у невролога на руках, тогда можно назначать определенный курс и схему лечения такому пациенту.
Как продлить жизнь пациенту?
В настоящее время к сожалению уровень медицины не дошел до того уровня, чтобы найти эффективные и быстрые методы борьбы с данным генетическим недугом.
Методы лечения до сих пор являются предметом поиска и изучения многих ученых. Однако для поддержания жизнеобеспечения таких больных принято применять паллиативное симптоматическое лечение.
Чтобы продлить срок жизни таких больных, назначают специальную иммунную терапию, которая может включать в себя различные дозировки препаратов Т-активина и гамма-глобулина.
При этом обязательной является постоянная высокая дозировка витаминных препаратов, которые вводятся комплексно для поддержания правильного функционирования всего организма.
Если при этом пациент с синдромом Луи-Бара имеет какое-то инфекционное заболевание, то его лечат в первую очередь интенсивной терапией, чтобы запустить процесс поддержания организма на должном уровне без излишних бактерий и вирусов.
В зависимости от нарушений, которые наблюдаются в организме, лекарственные препараты и их дозировки могут существенно меняться. Часто курс терапии дополняют противогрибковыми и противовирусными препаратами, а также сильными антибиотиками.
Если курс лечения длится довольно долго и имеет положительную тенденцию, то пациенту время от времени могут назначать введение иммуноглобулина и плазмы в организм.
Самовольное и беспричинное нагнетание психики по поводу несуществующих заболеваний — ипохондрический невроз. Что нужно сделать для терапии расстройства?Чем Джексоновская эпилепсия отличается от других видов заболевания. Методы диагностики и лечения рассматриваются здесь.
Реальные прогнозы
Поскольку синдром Луи-Бара довольно новый и полностью неизученный, то говорить о высоких шансах на лечение и тем более на выздоровление больного нельзя.
Патология имеет неблагоприятный прогноз, который в зависимости от разных факторов может как протекать на одном уровне много лет, так и стремительно катиться вниз.
Чаще всего симптом обнаруживают в глубоком детстве или при рождении ребенка. Средний возраст жизни таких детей составляет около 3 лет.
Если же симптомы проявились позже, то такие больные доживают максимум до 20-летнего возраста.
Чаще всего причиной их смерти является не само заболевание Луи-Бара, а полное уничтожение иммунитета и быстрое развитие онкологических образований по всему организму.
Синдром Луи-Бар
Наследственная патология, при которой наблюдаются мозжечковая атаксия, телеангиэктазии кожного покрова и конъюнктивы, а также недостаточность Т-клеточного иммунитета. Пациент страдает от частых респираторных инфекций. При синдроме Луи-Бар повышается вероятность образования неоплазий злокачественного характера. Диагноз устанавливают на основании анамнестической информации, клинических проявлений, физикального осмотр и дополнительных обследований. В рамках диагностики выполняют анализ крови на уровень иммуноглобулинов, ультразвуковое исследование, магнитно-резонансную томографию, фарингоскопию, риноскопию и рентгенографию. Больному назначают гамма-глобулин, препараты тимуса, витамины. Прогноз для жизни неблагоприятный. Большинство пациентов погибают до совершеннолетия. Причинами летального исхода чаще всего являются инфекционные осложнения и онкологические заболевания.
Причины
Патология образуется на фоне генетических нарушений, из-за которых развивается врожденная нейроэктодермальная дисплазия. Тип наследования недуга аутосомно-рецессивный. Чтобы синдром проявился, рецессивный ген должен быть сразу и у матери, и у отца. При патологии возникают дегенеративные изменения мозжечка: он теряет клетки Пуркинье и зернистые клетки. Возможны дегенеративные поражения зубчатого ядра мозжечка, черной субстанции, спиномозжечковых путей, задних столбов спинного мозга и некоторых отделов коры мозга. Отмечаются дефицит иммуноглобулинов А и Е, а также гипоплазия и аплазия тимуса. Именно из-за иммунных нарушений больные часто страдают инфекционными заболеваниями.
Симптомы
Первые признаки патологии возникают в период младенчества. Сперва развивается мозжечковая атаксия, проявляющаяся при ходьбе. Ребенку сложно удерживать равновесие, у него качается туловище и голова. Наблюдается интенционный тремор. Часто из-за атаксии малыш утрачивает способность ходить. Может отмечаться мозжечковая дизартрия – невнятная скандированная речь. Симптоматика дополняется гипотонией мышц, снижением либо выпадением рефлексов сухожилий, нистагмом, косоглазием и глазодвигательными нарушениями. В дошкольном возрасте появляются телеангиэктазии. Мелкие сосуды кожных покровов и конъюнктивы глаза расширяются и формируют «звездочки». Характерно снижение эластичности, сухость и уплотнение кожи. Заболевание может сопровождаться гипертрихозом, ранней сединой, сыпью напоминающей псориаз. Снижение иммунного ответа выражается частыми рецидивирующими инфекциями дыхательных путей. Больные страдают от хронического ринита, фарингита, бронхита, пневмонии, отита и синусита. Обычно данные болезни плохо поддаются лечению антибактериальными препаратами. На фоне частых легочных патологий формируются пневмосклероз и бронхоэктазы. Кроме того, одним из симптомов недуга является высокая вероятность образования злокачественных неоплазий. Чаще всего выявляют лейкемию и лимфому.
Диагностика
Больного консультирую специалисты неврологического, дерматологического, оториноларингического, офтальмологического, иммунологического, пульмонологического и онкологического профилей. Чтобы установить и подтвердить диагноз, доктор собирает анамнестическую информацию, анализирует клинические признаки, проводит физикальный осмотр и направляет пациента на дополнительные обследования. В рамках диагностики могут выполнять анализ крови на уровень иммуноглобулинов, ультразвуковое исследование, магнитно-резонансную томографию, фарингоскопию, риноскопию и рентгенографию. Патологию дифференцируют от наследственной геморрагической телеангиэктазии, болезни Гиппеля-Линдау, наследственной мозжечковой атаксии Пьера-Мари.
Лечение
Специфическое лечение все еще на стадии разработки. Медики проводят паллиативную симптоматическую терапию. Чтобы купировать иммунологические нарушения, назначают гамма-глобулин, высокие дозы витаминов, а также препараты тимуса. При любых инфекционных процессах проводят интенсивную антибактериальную, противовирусную или противогрибковую терапию. Возможно применение глюкокортикостероидов.
Профилактика
Первичной профилактики не существует. Парам, планирующим беременность, необходима консультация генетика. Пациентам, страдающим синдромом Луи-Бар, необходимо избегать инфекционных заболеваний.
симптомы, диагностика и лечение. Причины развития синдрома Луи-Бар
Атаксия-телеангиэктазия — сложное генетическое нейродегенеративное заболевание, которое может проявиться в раннем детстве. Заболевание характеризуется постепенным нарушение координации произвольных движений (атаксия), развитие красноватых поражений кожи и слизистой оболочки из-за постоянного расширения группы кровеносных сосудов (телеангиэктазии) и нарушениями функционирования иммунной системы (например, клеточного и гуморального иммунодефицита), что приводит к повышенной восприимчивости в верхних и нижних дыхательных путях. У людей с атаксией- телеангиэктазией также повышен риск развития определенных злокачественных новообразований, особенно рак лимфатической системы, кроветворных органов (например, лейкемия) или рак мозга.
Прогрессивная атаксия, как правило, развивается в грудном возрасте и поначалу может характеризоваться аномальным расхождением в движениях головы по отношению к туловищу. По мере прогрессирования заболевания это состояние приводит к невозможности нормально двигаться, а иногда даже и ходить к позднему детству или подростковому возрасту. Атаксия часто сопровождается затруднением в произношении слов вследствие нарушения речевого аппарата, а также нарушением способности координировать движения глаз, в том числе возникновением непроизвольных, быстрых, ритмичных движений глаз при попытке сосредоточиться на определенных предметах.
Кроме того, к 6-7 годам у ребенка может появится расширение мелких сосудов кожи, часто появляющиеся на открытых участках кожи, таких как мост носа, уши и определенные области конечностей, а также слизистые оболочки глаз.
Телеангиэктазия (стойкое расширение мелких сосудов) у ребенка, подобную картину можно заметить у пожилых лиц.
Ранним симптомом атаксии-телеангиэктазии является уменьшением мышечной координация, как правило, когда ребенок начинает ходить. Координация (особенно в области головы и шеи) становится нарушенной, и могут возникнуть непроизвольные сокращения мышц. В большинстве случаев психическое функционирование не затрагивается, и большинство детей в умственных способностях никак не отстают от детей без данного заболевания.
Видимые расширенные кровеносные сосуды обычно начинаются в глазах (глаза выглядят кровяным) в возрасте от трех до шести лет, хотя телеангиэктазия может проявится раньше. Эти пятна могут распространиться на веки, лицо, уши, и возможно, другие участки тела. Быстрое моргание глаз и движения, а также поворот головы могут развиваться постепенно. Иногда могут возникать носовые кровотечения. Аденоиды, миндалины и периферические лимфатические узлы могут развиваться аномально или не развиваться вовсе. Мышечная координация в области головы и шеи может быть постепенно нарушена, вызывая рефлексы кашля и проблемы с глотанием, дыханием.
Задержку роста можно объяснить дефицитом гормона роста. Преждевременное старение происходит примерно у девяноста процентов пораженных людей и характеризуется седыми волосами с сухой, тонкой, морщинистой или обесцвеченной кожей в подростковом возрасте.
Из-за нарушения иммунной системы больные с синдромом атаксии-телеангиэктазии подвергаются риску к хроническим или легочным инфекциям, повторяющимся случаям пневмонии и хронического бронхита.
Примерно у одного из трех пораженных людей развивается , как правило, рак определенных злокачественных новообразований, особенно лимфатической системы или лейкемии. Воздействие рентгеновских лучей, увеличивает частоту возможных опухолей.
В некоторых случаях может возникнуть легкая форма сахарного диабета . — это заболевание, при котором происходит недостаточная выработка гормона инсулина. Первичные симптомы могут проявиться в виде повышенной жажде и мочеиспускание, потерю веса, отсутствие аппетита и уста
Врожденный иммунодефицит: синдром Луи-Бар | Интернет-издание "Новости медицины и фармации"
Синдром Луи-Бар (врожденной атаксии-телеангиэктазии — А-Т) — врожденное иммунодефицитное состояние с преимущественным поражением Т-звена иммунитета, характеризуется аномальным развитием эмбриональных закладок и, по-видимому, неправильным взаимодействием эктодермы и мезодермы. Синдром Луи-Бар — это генетическое заболевание, которое наследуется по аутосомно-рецессивному типу. Впервые описано в 1941 г . D . Louis - Barr . Популяционная частота неизвестна. Соотношение полов: м : ж — 1 : 1 [1].
Иммунодефицит и хромосомная нестабильность являются маркерами А-Т ( Ataxia - Teteangiectasia Mutated ), что кодирует синтез одноименной киназы. Клетки пациентов с А-Т характеризуются повышенной чувствительностью к радиации, дефектами клеточного цикла, клинические же проявления и иммунологические нарушения имеют существенные различия, отмечаются повышенная частота развития злокачественных опухолей и спонтанная хромосомная нестабильность, хромосомные поломки, вовлекающие преимущественно 7-ю и 14-ю хромосомы [2, 3].
Известно, что клеточный цикл делится на 4 фазы: митоз (М) и синтез ДНК ( S ), разделенные двумя перерывами Gl и G 2. Последовательность клеточного цикла выглядит следующим образом: G 1 — S — G 2 — M . После воздействия ионизирующего излучения происходят двунитевые разрывы ДНК. Если происходит репапарация ДНК, то клеточный цикл восстанавливается, если нет — происходит гибель клетки путем апоптоза или развивается мутантный клон. В норме клеточный цикл при воздействии радиации может быть блокирован в двух критических точках — переходе из Gl -фазы в S -фазу и/или из G 2-фазы в М-фазу. При А-Т нарушен контроль клеточного цикла в критических точках. Двунитевые разрывы ДНК происходят в процессе рекомбинаций генов иммуноглобулинов и Т-клеточного рецептора. Процессы, напоминающие рекомбинацию генов иммуноглобулинов, происходят при созревании нейронов головного мозга. Очевидно, что именно с дефектами репарации ДНК в этих случаях связаны многие клинические и иммунологические проявления у больных с А-Т такие как нарушения со стороны синтеза иммуноглобулинов, функции половых органов и нервной системы [3, 4].
Клинические проявления А-Т могут существенно отличаться у разных больных. Прогрессирующая мозжечковая атаксия и телеэнгиэктазии присутствуют у всех, часто встречаются пятна «кофе с молоком» на коже. Склонность к инфекциям колеблется от очень выраженной до весьма умеренной. Очень высока частота развития злокачественных новообразований, преимущественно лимфоидной системы. Иммунологическими изменениями у больных с А-Т являются нарушения клеточного иммунитета в виде снижения количества Т-лимфоцитов, инверсии соотношения CD4+/CD8+ (в основном за счет снижения CD4+-клеток) и снижения функциональной активности Т-клеток. Со стороны концентраций сывороточных иммуноглобулинов наиболее характерным изменением является снижение или отсутствие IgA, реже выявляются концентрации иммуноглобулинов, близкие к норме, или дисиммуноглобулинемия в виде резкого снижения IgA, IgG, IgE и значительного повышения IgM. Характерно нарушение антителообразования в ответ на полисахаридные и белковые антигены. Методов излечения А-Т до настоящего времени не разработано. Больные нуждаются в паллиативной терапии неврологических и соматических расстройств. В случае выявления серьезных иммунологических изменений и/или хронических или рецидивирующих бактериальных инфекций показана антибактериальная терапия (длительность определяется тяжестью иммунодефицита и инфекции), заместительная терапия внутривенным иммуноглобулином, по показаниям — противогрибковая и противовирусная терапии [3, 4].
Клиническая характеристика. Заболевание начинается в раннем детстве и проявляется в первую очередь мозжечковой атаксией (100 %). Отмечаются качание головы и туловища, нарушение походки, интенционный тремор и хореоатетоз (90–100 %). Характерными изменениями глаз являются нарушение движения глазного яблока (80–90 %), нистагм (90–100 %) и косоглазие. В возрасте от 2 до 6 лет появляются телеангиэктазии на конъюнктиве и открытых участках тела, слизистой мягкого и твердого неба. Важным признаком синдрома являются хронические респираторные инфекции (синуситы и пневмонии, 60–80 %). Наблюдаются отставание в росте, пигментные пятна или участки депигментации на коже, склеродермия, гипотония мышц, гипорефлексия и дизартрия. У больных часто развиваются злокачественные новообразования, причем в 10–30 % поражается лимфоретикулярная система.
При патологоанатомическом исследовании обнаруживают аплазию или гипоплазию тимуса, уменьшение размеров лимфатических узлов и селезенки, признаки мозжечковой дегенерации, фиброзную дисплазию яичников. При А-Т имеет место нарушение В- и Т-клеточных систем иммунитета, которое выражается в отсутствии сывороточных иммуноглобулинов, главным образом IgA , но иногда IgG и IgE . При цитогенетическом исследовании лимфоцитов часто обнаруживают различные хромосомные аберрации и ломкость хромосом. Больные погибают от легочных инфекций либо от злокачественных новообразований [5].
На первое место в клинической картине выступает неврологическая симптоматика, поэтому болезнь вначале была описана как мозжечковая атаксия. В возрасте от 2 до 8 лет возникают телеангиэктазии, которые обычно располагаются на бульбарной конъюнктиве, между углом глаза и лимбом, и имеют вид красных извитых сосудов. Наблюдается аплазия вилочковой железы, гипоплазия (недоразвитие) лимфатических узлов, селезенки, групповых лимфатических фолликулов тонкой кишки, миндалин. У детей с синдромом Луи-Бар постоянно наблюдается гипоплазия (недоразвитие) или аплазия (полное отсутствие) небных миндалин. Лакуны миндалин недоразвиты. Шейные лимфатические узлы мелкие и не увеличиваются во время инфекций. Почти у всех детей с синдромом Луи-Бар выявляется хронический гнойный синусит, часто развивается отит [5].
Диагноз ставится на основе клинической картины, а также данных лабораторных показателей. У всех больных с синдромом Луи-Бар почти полностью отсутствуют Т-супрессоры. У части больных клетки не могут синтезировать IgA, что связано с отсутствием Т-хелперов. В крови обнаруживают a- и b-протеин. Патогенетическим методом лечения является аллотрансплантация неонатальной вилочковой железы. Назначают курс инъекций активных факторов вилочковой железы (Т-активина, тималина, тимацина и др.), систематически вводят нативную плазму и нормальный иммуноглобулин человека [5].
Под нашим наблюдением находилась девочка К., в клинику она поступила в возрасте 13 лет и 10 месяцев по поводу врожденного иммуннодефицитного состояния с атаксией (синдром Луи-Бар), хронической пневмонии, полисегментарного пневмосклероза, гнойного деформирующего эндобронхита, бронхоэктатической болезни в фазе обострения, правосторонней крупноочаговой пневмонии, осложненных генерализованным амилоидозом внутренних органов: печени с развитием цирроза и печеночной недостаточности, почек, селезенки, кишечника, анемии, кахексии.
При поступлении жалобы матери на желтушное окрашивание кожи, повторную рвоту, анорексию, общую слабость, исхудание. Из анамнеза известно, что родилась доношенной, с малым весом — 2 700 г , с оценкой по шкале Апгар 6–7 баллов. Находилась на естественном вскармливании, до года не болела. Со второго года жизни отмечались частые простудные заболевания, стало прогрессировать исхудание, перенесла повторную пневмонию. С 4 лет выявлена мозжечковая атаксия. Девочка консультирована в нашей клинике, в клинике г. Москвы диагностирован синдром Луи-Бар. С тех пор прогрессировали явления дистрофизации, атаксия, переносила повторные пневмонии. Диагностирована хроническая бронхоэктатическая болезнь. Неоднократно лечилась в стационаре. Последние 2 года жизни девочка не ходит, присоединились изменения со стороны печени и почек, связанные с амилоидозом. За 3 месяца до последней госпитализации находилась в клинике, диагноз подтвержден, получала комплексную терапию — антибиотики широкого спектра действия, дезинтоксикационную терапию, иммунотерапию. Состояние девочки стабилизировалось. Выписалась домой на поддерживающей дозе препаратов, улучшающих обменные процессы печени и почек. За 2 недели до поступления состояние пациентки резко ухудшилось, наросла желтуха, наблюдалась полная анорексия, появилась повторная рвота. Направлена в клинику.
При поступлении общее состояние тяжелое. Девочка резко дистрофизирована. Кожные покровы и склеры иктеричные, множественная «звездчатая» сыпь. На глазных яблоках выражен сосудистый рисунок. Заторможена, на вопросы отвечает вяло. Положение в постели горизонтальное, сидит с поддержкой. Видимые слизистые бледные. Язык розовый. Периферические лимфатические узлы мелкие, единичные до 0,5–1,0 см в диаметре, пальпируются подчелюстные. Пульс — 100. ЧД — 40. АД — 100/60 мм рт.ст. Над легкими перкуторно легочный звук, укорочен в нижних отделах, аускультативно дыхание жесткое, в нижних отделах ослабленное, выслушиваются единичные влажные мелкопузырчатые хрипы. Границы сердца расширены в поперечнике, левая — по передней аксиллярной линии. Тоны приглушены, ритмичны. Живот увеличен в объеме, при пальпации мягкий, асцита нет. Печень плотная, пальпируется на 4 см ниже реберной дуги, селезенка плотная, пальпируется на 5 см ниже реберной дуги у входа в малый таз. Мочится свободно. Стул оформлен, оправляется самостоятельно.
Лабораторные обследования
Анализ крови: Эр. — 2,9 Т/л, Н b — 90 г/л, Ц.П — 0,9, Лейк. — 8,2 Г/л, выражен анизоцитоз и пойкилоцитоз, п/я — 14 %, с/я — 20 %, л. — 64 %, м. — 2 %, СОЭ — 6 мм/час. Остаточный азот крови — 54,5 г/л. Холестерин крови — 4 мкмоль/л. АСТ — 0,35, АЛТ — 0,42. Общий билирубин крови — 84,8 ммоль/л, прямой — 74,2, непрямой — 10,6.
Сулемовая проба — 1,6. Общий белок крови — 64 г/л, альбумины — 46,7, гамма-глобулины — 19 %. Протромбин крови — 75 %.
Анализ мочи: белок — 0,86 г/л, Лейк. — 10–15, до 25 в п/зр., Эр. — 10 в п/зр., цилиндры гиалиновые — 1–2, зернистые — 1–2 в п/зр.
На рентгенограмме органов грудной клетки: легочная ткань умеренно вздута, особенно в нижних долях. Легочный рисунок усилен, расширен, справа в средней доле крупноочаговая инфильтрация легочной ткани без четких контуров. Синусы свободны. Сердце — в норме. ЭКГ: диффузное поражение миокарда. На основании анамнеза, объективных данных, клинического обследования и наблюдения выставлен вышеописанный диагноз.
Получала терапию: в/в капельно р-р Рингера, гемодез, плазма, корглюкон, лазикс, ампициллин в/м, ежедневно гамма-глобулин, сирепар, липоевая кислота, метионин, преднизолон, оксигенотерапия, диета № 7.
Несмотря на проводимую терапию, состояние девочки прогрессивно ухудшалось, нарастали явления печеночной и почечной недостаточности, уменьшался суточные диурез, последние дни до 300 г в сутки. В легких увеличилось количество хрипов, нарастала дыхательная и сердечная недостаточность. Через 18 дней после поступления в стационар состояние агонирующее, появилось носовое кровотечение, в кале примесь крови, стул дегтеобразный, появился печеночный запах. Проводимые реанимационные мероприятия эффекта не дали. При явлении печеночной с присоединением дыхательной и сердечной недостаточности девочка умерла на 20-й день пребывания в клинике.
Патологоанатомический диагноз
Основной : врожденное иммунодефицитное состояние с атаксией — синдром Луи-Бар. Хроническая пневмония. Полисегментарный пневмосклероз, гнойный деформирующий эндобронхит, бронхоэктатическая болезнь в стадии обострения, правосторонняя крупноочаговая пневмония.
Осложнения: генерализованный амилоидоз внутренних органов: печени с развитием цирроза и печеночной недостаточности, почек, селезенки, кишечника. Анемия. Кахексия.
Особенностью данного клинического случая можно считать редкую частоту встречаемости, характерную клиническую и лабораторную картину заболевания, медленное прогрессирование развития синдрома Луи-Бар, возраст пациентки.
Лечение синдрома Луи-Бар в Израиле | Детская больница «Сафра»
- Детская больница Сафра > Центр детской иммунологии > Cиндром Луи-Бар
Лечение атаксии-телеангиэктазии (синдром Луи-Бар)
Атаксия-телеангиэктазия (синдром Луи-Бар) представляет собой редкое наследственное иммунодефицитное заболевание, тем не менее, довольно часто встречающееся среди членов некоторых этнических групп Израиля.
Это прогрессирующее дегенеративное заболевание, протекающее с поражением различных систем организма. Непосредственно после рождения дети, страдающие атаксией-телеангиэктазией, не проявляют никаких признаков заболевания.
Как правило, первые симптомы, заключающиеся в нарушении равновесия ( баланса) и речи (атаксии), проявляются на втором году жизни и свидетельствуют о прогрессирующем поражении нервной системы.
Проявление данных симптомов вызвано нарушением координации произвольных мышечных движений вследствие дегенеративных изменений в мозжечке, постепенно приводящих к полной потере контроля над мышцами и к тяжёлой инвалидности.
В процессе усугубления атаксии, дети утрачивают способность писать, разговаривать, а впоследствии и читать по причине утраты контроля над иннервацией мышц, отвечающих за движение глазных яблок.
Характерным симптомом заболевания является также появление телеангиэктазий (расширенных капилляров) на конъюнктиве глаз, коже лица и мочек ушей, открытых для солнечного света. Данные симптомы возникают, как правило, уже после развития атаксии, но во многих случаях их характерный вид способствует диагностике заболевания и объясняет его название.
Поражение иммунной системы
У большинства (≈70%) детей, страдающих атаксией-телеангиэктазией, наблюдается поражение иммунной системы, предрасполагающее к возникновению частых инфекционных заболеваний, в частности пневмонии, отитов и др. и к развитию онкологических заболеваний, особенно различных видов лейкемии и лимфомы.
Предрасположенность к рецидивирующим респираторным инфекциям
Во многих случаях, рецидивирующие респираторные инфекции несут настоящую угрозу для жизни пациентов, страдающих атаксией-телеангиэктазией. Из-за недостатка иммуноглобулинов IgA и IgE, являющихся естественными звеньями в борьбе с инфекцией, подобные пациенты крайне подвержены инфекционным заболеваниям лёгких, не поддающимся стандартной антибактериальной терапии.
Сочетание иммунодефицита и прогрессирующей атаксии предрасполагает таких пациентов к развитию пневмонии, которая является одной из наиболее распространённых причин их смерти.
Предрасположенность к развитию злокачественных новообразований
Случаи злокачественных заболеваний кроветворной системы среди детей, страдающих атаксией-телеангиэктазией, встречаются в 1000 раз чаще, чем среди прочих групп населения. В частности, такие заболевания, как лимфома и лейкемия являются наиболее распространёнными видами онкопатологии среди таких пациентов, хотя наблюдается учащение случаев возникновения всех видов злокачественных опухолей.
В то же время, другим аспектом заболевания, является повышенная чувствительность к действию ионизирующей радиации, что исключает возможность проведения радиотерапии, обычно применяемой при лечении онкологических заболеваний.
Другие проявления атаксии-телеангиэктазии
Другими проявлениями атаксии-телеангиэктазии, наблюдаемыми у некоторых пациентов, могут быть лёгкая форма сахарного диабета, раннее поседение волос и дисфагия (нарушение глотания), ведущая к затруднительному приёму пищи, слюноотделению и замедлению роста.
Следует отметить, что, несмотря на то, что атаксия-телеангиэктазия является мультисистемным заболеванием, ведущим к прогрессирующей потери трудоспособности, оно никак не отражается на умственном развитии ребёнка.
Частота заболевания
Данным заболеванием страдают в равной степени дети обоих полов, без учёта национальности, экономического и географического положения или уровня образования.
По оценкам эпидемиологов, заболевание встречается один раз на 40000 новорожденных.
Считается, что во многих случаях, особенно при смерти в раннем возрасте, заболевание остаётся нераспознанным. Таким образом, оно может оказаться более распространённым.
Доступные виды лечения
Поскольку атаксия-телеангиэктазия является очень редким заболеванием, исследовательских данных относительно его фармакологической терапии крайне мало. Существующее лечение направлено исключительно на частичное облегчение симптомов по мере их появления.
При лечении применяются физиотерапия, трудовая терапия и занятия с логопедом для улучшения адаптации к окружающему миру и социальной среде. Также используются инъекции гамма-глобулина для повышения иммунитета пациентов и витаминотерапия в высоких дозировках, приносящие некоторое улучшение.
Клиника по лечению атаксии - телеангиэктазии (синдром Луи-Бар)
Лечащий персонал клиники по лечению синдрома Луи-Бар
ЛУИ-БАР СИНДРОМ - это... Что такое ЛУИ-БАР СИНДРОМ?
- ЛУИ-БАР СИНДРОМ
- (синдром Бодера – Седвика, описан французским врачом D. Louis-Bar, позднее – американскими врачами E. Boder, R. P. Sedgwick; синоним – атаксия-телеангиэктазия) – наследственное заболевание, начинающееся в раннем детском возрасте с неврологической симптоматики – мозжечковой атаксии с нарушением походки, дрожания головы и туловища, хореоатетоза, интенционного тремора, нарушения движения глазных яблок. В возрасте 2–6 лет появляются асимметричные телеангиэктазии на открытых участках тела, слизистой оболочки ротовой полости; наблюдаются отставание в росте; нарушение пигментации кожи; хронические бронхо-легочные инфекции, вызванные комбинированным (гуморальный и клеточный) иммунодефицитом, причем максимально снижен или отсутствует сывороточный иммуноглобулин А, иногда G и E; у большинства больных наблюдаются нарушение толерантности к глюкозе или сахарный диабет. Тип наследования – аутосомно-рецессивный. При цитогенетическом исследовании лимфоцитов выявляют хромосомную нестабильность (повышенный уровень спонтанных хромосомных аберраций и ломкость хромосом). При патологоанатомическом исследовании находят гипоплазию или аплазию вилочковой железы, лимфатических узлов, селезенки, фиброзную дисплазию яичников и др. Диагноз может быть подтвержден ДНК-диагностикой, в том числе пренатально (биопсия ворсинчатого хориона, амниоцентез, кордоцентез). Лечение: иммунокоррекция, диетотерапия, паллиативная терапия неврологических расстройств, антибактериальная, противовирусная, противогрибковая терапия. Продолжительность жизни часто сокращена – больные погибают от злокачественных новообразований, чаще всего лимфоретикулярной системы, или от осложненных легочных инфекций.
D. Louis-Bar. Sur un syndrome progressif cormprenant des telangiectasies capillaires cutanees et conjonctivales symetriques, r` disposition naevo?de et des troubles cerebelleux. Confinia Neurologica, 1941 ; 4: 32–42.
E. Boder, R. P. Sedgwick. Ataxiatelangiectasia: a familial syndrome of progressive cerebellar ataxia, oculcutaneous telangiectasia and frequent pulmonary infection. Pediatrics, 1958; 21(4): 526–554.
Энциклопедический словарь по психологии и педагогике. 2013.
- ЛСД-25
- Лукавить
Смотреть что такое "ЛУИ-БАР СИНДРОМ" в других словарях:
Луи-Бар синдром — (D. Louis Bar, совр. франц. врач; син. атаксия телеангиэктазия) наследственная болезнь из группы факоматозов, характеризующаяся медленно развивающимися мозжечковыми расстройствами, симметричными телеангиэктазиями, особенно на конъюнктивах, коже… … Большой медицинский словарь
синдром Луи-Бар — ataxia teleangiectasia, Louis Bar syndrome атаксия телеангиэктазия, синдром Луи Бар. НЗЧ, проявляющееся в виде одной из форм атаксии (нарушения координации движений), а также характеризующееся поражениями кожи, дефицитом иммунной системы (резко… … Молекулярная биология и генетика. Толковый словарь.
Синдром Луи-Бар — Син.: Атаксия телеангиэктазия. Наследственное заболевание из группы факоматозов. Характеризуется медленно развивающимися расстройствами функций мозжечка, симметричными телеангиэктазиями на конъюнктиве глаз, коже лица и шеи, позже появляются… … Энциклопедический словарь по психологии и педагогике
Атакси́и — (греч. ataxia отсутствие порядка, беспорядочность; синоним инкоординация) нарушение координации (согласованности действия) различных мышц, проявляющееся расстройством статических функций и целенаправленных движений. Для целесообразного выполнения … Медицинская энциклопедия
Гемобластозы — МКБ 10 C … Википедия
атаксия-телеангиэктазия — см. Луи Бар синдром … Большой медицинский словарь
Атакси́я-телеангиэктази́я — см. Луи Бар синдром … Медицинская энциклопедия
Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни (врождённые иммунодефициты). Однако менее выраженные генетические дефекты… … Википедия
Иммунопатология — I Иммунопатология (иммуно[логия] (Иммунология) + Патология раздел иммунологии, изучающий поражение иммунной системы при различных заболеваниях. Отсутствие одной из нескольких субпопуляций клеток иммунной системы проявляется как врожденное… … Медицинская энциклопедия
атаксия-телеангиэктазия — синдром Луи Бар НЗЧ, проявляющееся в виде одной из форм атаксии (нарушения координации движений), а также характеризующееся поражениями кожи, дефицитом иммунной системы (резко снижено содержание иммуноглобулина А), расстройствами речи и др.;… … Справочник технического переводчика