Синдром свайера что это такое
Синдром Свайера - причины, симптомы, диагностика и лечение
Синдром Свайера – это нарушение формирования пола, характеризующееся кариотипом 46XY, врождённой дисгенезией гонад при первично сформированных прочих женских гениталиях – влагалище, матке, фаллопиевых трубах. К признакам патологии относятся первичная аменорея, маскулинное телосложение, половой инфантилизм. Диагноз выставляется на основании анамнестических данных, результатов общего и гинекологического осмотра, интроскопических методов исследования органов малого таза, гормонального и молекулярно-генетического анализа. Лечение состоит из двух этапов – удаления неразвитых половых желёз и длительного применения заместительной гормональной терапии.
Общие сведения
Синдром Свайера (полную, или «чистую» дисгенезию гонад) можно кратко охарактеризовать как женский фенотип при мужском генотипе. Заболевание названо по имени британского эндокринолога Джералда Свайера, описавшего его в 1955 году как случай мужского псевдогермафродитизма. Полная форма дисгенезии является несиндромальной (не сопровождается экстрагенитальными пороками развития), исключает двойственность полового развития (наличие мужских первичных половых признаков наряду с женскими), психологическое развитие происходит по женскому типу. Врождённая патология встречается в одном случае на 20 000 индивидов с мужским кариотипом и регистрируется чаще иных форм XY-дисгенезии гонад.
Синдром Свайера
Причины
Этиология синдрома Свайера изучена недостаточно. На сегодняшний день известно, что чаще всего возникновение патологии связано с отсутствием или мутацией гена SRY, расположенного на коротком плече хромосомы Y и большей частью отвечающего за контроль формирования тестикул. Судя по наблюдениям семейных случаев заболевания, возможна причастность пока неизвестных X-сцепленных или аутосомных генов.
Факторы риска тоже до конца не установлены. Кроме общих мутагенных воздействий (ионизирующего излучения и интоксикаций, вирусных инфекций, несбалансированного или пониженного питания) и уже упомянутого наследственного отягощения предполагается, что вероятность патологии может находиться в прямой зависимости от возраста отца. Чаще всего проследить связи между каким-либо воздействием на организм во время гестации и развитием синдрома Свайера не удаётся.
Патогенез
Формирование репродуктивных органов происходит из мюллерова протока у женщин и вольфова – у мужчин. У эмбриона с мужским генотипом синтез мужских стероидных гормонов клетками Лейдига в эмбриональных семенниках обусловлен воздействием материнского хорионического гонадотропина. Клетки Сертоли стимулируют дифференцировку клеток Лейдига и прочих, продуцируют антимюллеров гормон, способствующий атрофии мюллерова протока. Их нормальная деятельность обуславливает развитие индивида мужского пола – с адекватной дифференцировкой тестикул, атрофией мюллерова протока.
Выраженные сбои этого механизма приводят к формированию из бипотентных зачатков женских репродуктивных органов, развитие которых не требует столь сложной регуляции. Созревание мужского эмбриона контролируется геном SRY. При его отсутствии или мутации нарушается деятельность клеток Сертоли, дифференцировка гонад не происходит, что влечёт развитие синдрома Свайера – фенотипически женского организма без полноценных яичников, способных в дальнейшем стимулировать развитие вторичных половых признаков, но с бесполезными, склонными к малигнизации зачатками желёз.
Симптомы
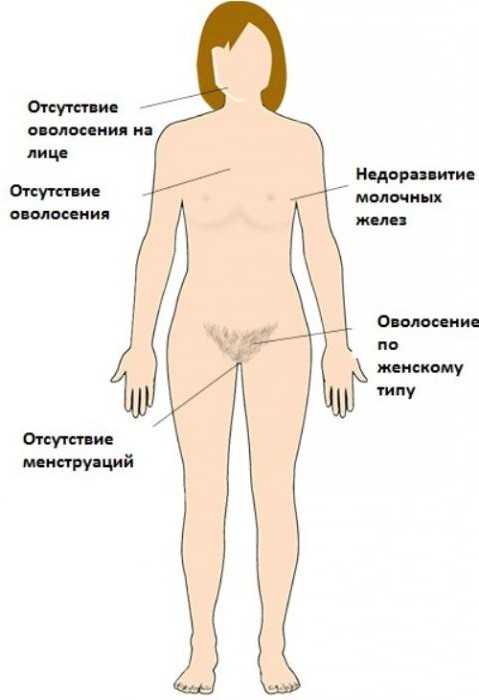
В допубертатном периоде патология протекает без каких-либо субъективных проявлений. В период пубертата для синдрома Свайера характерно отсутствие признаков полового созревания. Может отмечаться лишь скудное оволосение в области лобка и подмышек, однако нередко отсутствует и оно. Менархе не наступает, молочные железы не развиваются или выражены очень слабо. Тип телосложения при овариальной дисгенезии мужской – с широкими плечами, объёмной грудной клеткой, узким тазом.
У женщин с синдромом Свайера чаще наблюдается нормальный или выше среднего рост, развитая мускулатура, «тяжёлая» нижняя челюсть. Иногда возникает незначительная гипертрофия клитора, хотя обычно наружные гениталии несколько недоразвиты. Больные жалуются на первичное бесплодие, ощущение дискомфорта или боли ввиду недостаточного развития вагины при половом акте или гинекологическом осмотре.
Осложнения
Частым (у 20-60% пациенток) осложнением синдрома Свайера является развитие опухолей, по большей части исходящих из рудиментарного полового тяжа, которым, по сути, представлены дисгенетичные гонады – дисгермином, андробластом. Эти неоплазии нередко имеют злокачественный характер, возникают с обеих сторон (синхронно или метахронно), регистрируются в раннем репродуктивном, подростковом и детском возрасте.
К последствиям нелеченного синдрома Свайера также можно отнести раннее появление патологий, обусловленных дефицитом эстрогена у женщин – остеопороза, сердечно-сосудистых заболеваний (артериальной гипертензии, ишемической болезни сердца). Кроме того, для многих больных диагноз становится источником тяжёлого психологического страдания по поводу «утраты женственности», а у замужних женщин – и боязни распада семьи.
Диагностика
Диагностика синдрома Свайера осуществляется гинекологом при участии медицинского генетика. Чаще всего диагноз устанавливается в 14-15 лет при обращении к врачу по поводу отсутствия полового развития, иногда – позднее, по причине бесплодия. В некоторых случаях первым признаком заболевания и основанием для углублённого исследования является опухолевое образование рудиментарных половых желёз, случайно обнаруженное врачом или самой больной. Диагностические мероприятия включают:
- Клинический осмотр. В ходе общего осмотра выявляется отсутствие основных вторичных женских половых признаков на фоне нормального или ускоренного роста. При гинекологическом осмотре обнаруживаются нормально сформированные, но недоразвитые женские наружные половые органы. Косвенным подтверждением диагноза является наличие кровных родственниц с полной или частичной дисгенезией гонад.
- Лучевые методы исследования. Наиболее доступным и информативным методом диагностики является гинекологическое УЗИ. Ультрасонография позволяет обнаружить такие характерные для патологии объективные признаки, как гипоплазию матки, извитые, недоразвитые маточные трубы, тяжевидные «яичники» (иногда – с новообразованиями). В сомнительных случаях назначается МРТ.
- Гормональный анализ. Характерным признаком синдрома Свайера считается значительное повышение гонадотропных (фолликулостимулирующего, лютеинизирующего) гормонов в сыворотке крови, снижение уровня эстрадиола. Гестагеновая функциональная проба отрицательная, циклическая (эстроген-гестагенная) гормональная проба положительная.
- Генетическое тестирование. Применяется с целью верификации диагноза. Цитогенетическое исследование выявляет мужской кариотип (46, XY), а молекулярно-генетическое – отсутствие или повреждение гена SRY. При невозможности генетического обследования назначается гистологическое исследование биоптата яичников. Диагноз подтверждает соединительнотканная структура с отсутствием фолликулов.
Синдром Свайера дифференцируют с другими формами дисгенезии гонад – синдромом Шерешевского-Тёрнера, мозаицизмом 45,X/46,XY,
Синдром Свайера и женская гонадальная дисгенезия
Содержание :
- Строение тела при синдроме Свайера
- Причины патологии
- Клиническая картина синдрома Свайера
- Диагностика болезни
- Лечение
Синдром Свайера – (полная, или «чистая» дисгенезия гонад) можно кратко охарактеризовать как женский фенотип при мужском генотипе. Это врожденное заболевание, которое возникает вследствие сбоя в хромосомах. Больной внешне выглядит как девушка (женский фенотип), но при исследовании полового хроматина определяются X и Y хромосомы, то есть мужской кариотип. Данную патологию также часто называют инверсией пола.
Строение тела при синдроме Свайера
Внешне невозможно отличить больную девушку от здоровой. Люди, страдающие синдромом Свайера, имеют женское телосложение, нормальный рост и сформированные внешние половые органы. Зачастую у них физиологически правильно развита матка и фаллопиевы трубы, но иногда бывает их гипоплазия. Яичники сильно видоизменены, они не могут выполнять свои функции. При обследовании на их месте обнаруживаются тяжи из соединительной ткани, могут присутствовать железистые включения. Такие люди полностью стерильны, то есть у них нет фолликулов с яйцеклетками, поэтому забеременеть природным путем они не могут.
Причины патологии
За развитие плода соответственно мужскому полу отвечает определенный белок. Он связывается с генами, которые несут информацию о половой принадлежности особи. Но чтобы этот белок правильно действовал, нужен ген Sexdetermining region Y, который будет его кодировать. Мутации SRY приводят к неправильной дифференциации клеток Сертоли, из-за этого происходит патологическое развитие семенных канальцев. Несмотря на мужской кариотип, остальные органы плода формируются женскими. Этот процесс является причиной появления дисгенезии гонад.
Клиническая картина синдрома Свайера
До периода полового созревания, симптомы синдрома Свайера неярко выражены. Но со взрослением девочки можно заметить некоторые особенности:
- недостаточный рост волос в подмышечных впадинах и на лобковой области;
- слаборазвитые грудные железы;
- инфантилизм матки, реже гипоплазия влагалища;
- интерсексуальное или евнухоподобное телосложение;
- атрофия слизистой оболочки в половых органах;
- недоразвитие клитора и половых губ;
- генитальный инфантилизм.
Диагностика болезни
В основном диагностируется заболевание у девочек в 15-16 лет. Это связано с тем, что они приходят к гинекологу с жалобами на задержку менархе (первой менструации). Иногда заболевание обнаруживается вследствие малигнизации (перерождения в рак) дисгенетических гонад.
Постановка диагноза базируется на данных осмотра, УЗИ, гистеросальпингографии и прочего. Но подтвердить болезнь можно только с помощью исследования полового хроматина, в котором определяется мужской кариотип при женском фенотипе.
Лечение
В первую очередь проводят овариоэктомию – удаление яичников, чтобы избежать их перерождение в раковые опухоли. После операции назначают гормональные препараты в качестве заместительной терапии. Это помогает развиться вторичным половым признакам, а также не дает возникнуть остеопорозу. Если удаление женских половых желез было произведено в детстве, то гормоны начинают давать только в подростковом возрасте. При своевременном обнаружении и лечении данной проблемы можно положительным образом повлиять на качество и продолжительность жизни при синдроме Свайера.
Женщины с синдромом Свайера не имеют своих яйцеклеток, но при достаточно развитой матке, они могут выносить и родить здорового ребенка. В таких случаях, беременность достигается путем экстракорпорального оплодотворения с введением донорских ооцитов в полость матки пациентки.
Синдром Свайера: причины, симптомы, диагностика, лечение
До того момента, когда ребенок входит в период половозрелости, симптоматика синдрома Свайера клинически отсутствует. Только по ходу взросления обнаруживаются соответствующие патологические признаки:
- отсутствие или малая выраженность волосяного покрова в зоне лобка и подмышек;
- малая развитость молочных желез;
- «детская» матка, иногда на фоне влагалищной гипоплазии;
- интерсексуальная вариация телосложения;
- атрофические процессы в слизистой ткани половых органов;
- недоразвитость внешних половых органов.
Половой зрелости у пациенток, болеющих синдромом Свайера, не происходит: в организме не вырабатываются необходимые гормоны – эстрогены. У большинства таких женщин полностью отсутствует месячный цикл, либо имеются незначительные менструальноподобные выделения.
В более зрелом возрасте у пациенток может сочетаться синдром Свайера и лимфангиолейомиоматоз – это редкая полисистемная болезнь, с нарастающими кистозными деструктивными изменениями легочной ткани. Поражается лимфатическая система, формируются ангиомиолипомы в органах брюшной полости. Патология проявляется спонтанным пневмотораксом, нарастающей одышкой, эпизодическим кровохарканьем. Подобные случаи единичны, однако на них стоит обращать внимание.
Особенности тела с синдромом Свайера
По внешним характеристикам не получится отличить здоровую девочку от больной синдромом Свайера. Пациентам, болеющим синдромом, присуще сложение тела в пределах нормы, адекватные показатели роста и сформированная наружная половая сфера. У многих из них обнаруживается физиологически нормальное развитие матки и придатков, однако часто присутствует гипоплазия. Наблюдается дисфункция яичников на фоне явного видоизменения органов.
По достижению половозрелого возраста наружные признаки синдрома у некоторых пациентов проявляются более выраженно. Наблюдается интерсексуальный или евнуховидный тип телосложения: широкие плечи, узкий таз, высокий рост, массивная нижняя челюсть, объемные мышцы.
Во время обследования вместо яичников иногда обнаруживают соединительнотканные элементы, включения из железистой ткани. Пациенты с синдромом Свайера являются стерильными – то есть, у них не происходит созревания фолликулов, отсутствуют яйцеклетки, они не могут зачать ребенка естественным способом.
[35], [36], [37], [38], [39]
особенности заболевания и возможности лечения
Синдром Свайера – это довольно редко встречающееся врожденное заболевание, развитие которого свидетельствует о нарушении структуры игрек-хромосомы (речь идет об отсутствии определенного гена или его изолированой мутации).
Причины развития заболевания
Как правило, непосредственной причиной возникновения данного заболевания является точечная мутация определенного гена, находящегося в коротком плече игрек-хромосомы, или полная утрата этого гена. Данный участок хромосомы отвечает за синтез белка, принимающего участие в развитии пола эмбриона по мужскому типу. В результате, поскольку воздействие мужских половых гормонов отсутствует, плоду остается единственный вариант – формироваться по женскому типу. В результате родившийся ребенок имеет женский фенотип при кариотипе "XY".
Патогенез
Мутации или отсутствие гена SRY ведет к сбою в дифференциации клеток Сертоли, и, как следствие, к недоразвитию семенных канальцев.
В результате, невзирая на «мужской» кариотип XY, половые органы плода закладываются и формируются по женскому типу.
Клинические проявления
Вплоть до начала пубертатного периода признаки синдрома Свайера внешне практически не выражены. И лишь по мере взросления девочки начинают проявляться определенные особенности:
- Недостаточность оволосения подмышечных впадин и области внешних половых органов.
- Недостаточное, слабое развитие молочных желез.
- Различной степени недоразвитие, инфантилизм матки.
- Гипоплазия влагалища (встречается реже).
- Неярко выраженные вторичные половые признаки – «евнухоподобный» или интерсексуальный тип телосложения.
- Гипотрофия или атрофия слизистой оболочки половых органов.
- Недоразвитие наружных половых органов (половых губ и клитора).
- Генитальный инфантилизм.
- В ряде случаев наблюдается чрезмерно активный рост тела и его отдельных частей: нижней челюсти, плечевого пояса (в результате чего формируются широкие плечи), мышечной массы.
- Наступление полового созревания у девушек с синдромом Свайера невозможно ввиду отсутствия в их организме эстрогенов.
- Полная стерильность.
Диагностика
В подавляющем большинстве случаев заболевание диагностируется в возрасте 15-16 лет, в период полового созревания, когда становится очевидно, что у пациентки не выражены вторичные половые признаки.
В это же время девушки, имеющие такую мутацию, достигнув этого возраста, начинают обращаться к гинекологу с жалобами на задержку наступления менструаций.
Иногда диагностика осуществляется в результате дисплазии и малигнизации недоразвитых гонад.
Постановка диагноза «синдром Свайера» основывается на следующих факторах:
- Физикальное обследование пациентки.
- Ультразвуковое обследование.
- Гистеросальпингография.
Однако подтверждение диагноза возможно лишь при помощи исследования полового хроматина, которое выявляет наличие мужского кариотипа при женском фенотипе.
Возможности лечения
Лечение синдрома Свайера осуществляется в нескольких направлениях.
- В первую очередь производится удаление яичников – ввиду высокой вероятности преобразования их в злокачественные опухоли.
- После овариоэктомии назначается заместительное лечение при помощи гормональных препаратов. Это способствует развитию вторичных половых признаков.
- В случае, когда матка развита достаточно, существует возможность вынашивания и рождения здорового ребенка (беременность наступает в результате экстракорпорального оплодотворения).
Данное заболевание следует отличать от сходного по названию синдрома Свайера-Джеймса-МакЛеода. Это состояние, как и сходный по проявлениям лимфангиолейомиоматоз, является патологией, поражающей легочную ткань. Синдром Свайера и лимфангиолейомиоматоз – разные заболевания.
Синдром Свайера - Здоровье и Стиль Жизни Женщин
Синдром Свайера представляет собой чистую форму дисгенезии гонад. При синдроме Свайера наблюдается наличие женского фенотипа при нормальном мужском кариотипе (46 XY). Подобное состояние называется инверсией пола.Частота XY дисгенезии гонад составляет 1 на 30000 человек.
Синдром Свайера – особенности
Женщины с синдромом Свайера имеют телосложение по женскому типу, нормально развитую или гипоплазированную матку и маточные трубы. Женские половые железы (яичники) практически отсутствуют и представлены соединительнотканными тяжами с незначительными включениями железистой ткани и овариоподобной стромой без фолликулов. Дисгенетические гонады подвержены озлокачествлению. Наружные половые органы также развиты по женскому типу.
Синдром Свайера – диагностика
Диагностируют Синдром Свайера, как правило, в период полового созревания. У девочек не происходит нормального полового развития, не наступает менархе (первая в жизни менструация). Реже, поводом обращения к врачу является наличие злокачественных образований, происходящих из дисгенетических гонад. Диагноз синдрома Свайера ставится после проведения генетического исследования и определения мужского кариотипа (46 ХУ) у девочки.
Синдром Свайера – причины
Наиболее частой причиной формирования синдрома Свайера являются микроструктурные перестройки У-хромосомы с утратой гена SRY (Sexdetermining region Y), а также точечные мутации данного гена. Ген SRY локализован на коротком плече Y хромосомы и кодирует белок, который связывается с генами, определяющими развитие пола плода по мужскому типу. Мутации в гене SRY приводят к синтезу функционально неполноценного белка и к нарушению дифференцировки клеток Сертоли. В результате происходит нарушение формирования семенных канальцев в развивающихся бипотенциальных гонадах плода. Подобные нарушения являются причиной формирования дисгенетических гонад. Развитие остальных органов половой системы происходит по женскому типу, несмотря на наличие Y-хромосомы в кариотипе.
Синдром Свайера – симптомы
Синдром Свайера , как правило сопровождается следующими клиническими признаками:

Что такое Синдром Свайера?
- Половой инфантилизм у женщин с нормальным или высоким ростом;
- Отсутствие соматических аномалий развития;
- Диспластичное телосложение по интерсексуальному или евнухоидному типу;
- Скудное оволосение в лобковой и подмышечной областях;
- Недоразвитие молочных желёз;
- Возможно недоразвитие наружных половых органов;
- Недоразвитие матки, а иногда и влагалища;
- Атрофичная слизистая оболочка половых органов.
Синдром Свайера – лечение
Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XУ дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, с целью достижения нормального развития вторичных половых признаков и предотвращения развития остеопороза. У женщин с XY дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях возможно вынашивание беременности, полученной в результате проведения экстракорпорального оплодотворения (ЭКО) с использованием донорских яйцеклеток.
Соответствующие ссылки из информации о женщинах / Google
Синдром свайера - XY gonadal dysgenesis
Синдром Swyer или синдром свайера , представляет собой тип гипогонадизма у человека , которого кариотип 46, XY. Человек внешне женщина с стрик гонад , и если его не лечить, не будет испытывать половое созревание . Такие гонады , как правило , удалены хирургическим путем ( так как они имеют значительный риск развития опухолей) и типичный лечение будет включать в заместительной гормональной терапии . Синдром был назван Джеральд Swyer, эндокринолога, базирующегося в Лондоне .
генетика
Генетические ассоциации синдрома Swyer включают в себя:
| Тип | OMIM | Ген | годограф |
|---|---|---|---|
| 46, синдром свайер, полный, SRY, связанный с | 400044 | SRY | Yp11.3 |
| 46, синдром свайер, полный или частичный, связанный с DHH | 233420 | DHH | 12q13.1 |
| 46, синдром свайер, полный или частичный, с или без недостаточности надпочечников | 612965 | NR5A1 | 9q33 |
| 46, синдром свайера, полная, CBX2 связанных | 613080 | CBX2 | 17q25 |
| 46, синдром свайер, полный или частичный, с удалением 9p24.3 | 154230 | DMRT1 / 2 | 9p24.3 |
Семь других генов были идентифицированы с возможными ассоциациями, которые пока еще менее ясно поняты.
Чистая дисгенезия гонад
Есть несколько форм дисгенезии гонад . Термин «чистая дисгенезия гонады» (PGD) используется для описания условий с нормальными наборами половых хромосом (например, 46, XX или 46, XY), в отличие от тех , чьих дисгенезии гонад результатов из -за отсутствие всех или части второго половые хромосомы. Последняя группа включает в себя те , с синдромом Тернера (т.е., 45, X) и его варианты, а также тех , с смешанной дисгенезии гонад и смеси клеточных линий, некоторые , содержащие Y - хромосому (например, 46, XY / 45, Х) ,
Таким образом , синдром Swyer упоминается как PGD, 46, XY и XX дисгенезии гонад , как PGD, 46, XX. Люди с PGD имеют нормальный кариотип , но может иметь недостатки конкретного гена на хромосоме.
патогенез
Первый известный этап половой дифференцировки нормального XY плода является развитие семенников . Ранние этапы формирования яичек во втором месяце беременности требует действие нескольких генов , из которых один из самых ранних и наиболее важным является SRY , то область Y - хромосомы секс-определения. Мутации SRY счета для многих случаев синдрома Swyer.
Когда такой ген является дефектным, то безразличные гонады не дифференцируются в семенниках в (генетически мужского пола) плода XY. Без яичек, не тестостерон или antimüllerian гормон (АМГ) не производится. Без тестостерона , то Вольфианская каналы не развиваются, поэтому не образуются внутренние органы мужчины. Кроме того , отсутствие тестостерона означает , что ни один дигидротестостерон не образуется и , следовательно , наружные половые органы не в состоянии virilize , в результате чего в нормальных женских половых органах. Без АМГА, в мюллеровых протоках развиваются в нормальные внутренние женские половые органы ( матка , маточные трубы , шейки матки , влагалище ).
Ребенок , который, по- видимому девочка рождаются и нормально в большинстве анатомических отношений , за исключением , что у ребенка есть нефункциональные хронограммы гонада вместо яичников или семенников. Как яичники девочек обычно не производят важные изменений тела до полового созревания , дефект репродуктивной системы , как правило , остается незаподозренным до полового созревание не не происходит у людей с синдромом Swyer. Они , кажется, нормальные девушки , и , как правило , считается так.
диагностика
Из - за неспособности Подряд гонад производить половые гормоны (как эстрогены и андрогены ), большинство вторичных половых признаков не развиваются. Это особенно верно в отношении эстрогенных изменений , таких как развитие груди, расширение таза и бедра, и менструаций . Поскольку надпочечники могут сделать ограниченное количество андрогенов и не страдают от этого синдрома, большинство из этих людей разовьет лобковые волосы, хотя она часто остается мало.
Оценка задержки полового созревания обычно показывает повышение гонадотропинов , что свидетельствует о том , что гипофиз обеспечивает сигнал для полового созревания , но яичники не в состоянии ответить. Следующие этапы оценки , как правило , включают в себя проверку кариотипа и формирование изображения таза. Кариотип показывает XY хромосомы и визуализация демонстрирует наличие матки , но нет яичников (Подряд гонады обычно не рассматриваются большинство изображений). Хотя кариотип XY может также указывать на человек с полным синдромом нечувствительности андрогенов , отсутствие молочных желез, а также наличие матки и лобковых волос исключает возможность. На этом этапе, как правило , возможно врач поставить диагноз синдрома Swyer.
Связанные условия
Синдром Swyer представляет собой один фенотипический результат отказа гонад развиваться должным образом, и , следовательно , является частью класса условий , называемых дисгенезии гонад . Есть много форм дисгенезии гонад.
Синдром Swyer является примером состояния , в котором внешне однозначно женское тело несет dysgenetic, атипичный, или ненормальные гонады. Другие примеры включают в себя полный синдром нечувствительности андрогена , частичные Х - хромосомы делеции, липидный врожденной гиперплазии коры надпочечников и синдром Тернера .
лечение
При диагностике, эстроген и прогестаген терапии , как правило , начато, способствуя развитию женских характеристик.
Последствия стрик гонад к человеку с синдромом Swyer:
- Половые не могут сделать эстроген, так что грудь не будет развиваться , и матка не будет расти и менструаций до эстрогена не вводят. Это часто дается трансдермально.
- Половые не могут сделать прогестерон, поэтому менструации не будут предсказуемы до прогестина не вводят, как правило , в виде таблеток.
- Половые не могут производить яйца так зачатие детей , естественно, не представляется возможным. Женщина с маткой и яичниками , но без женских гамет могут стать беременным путем имплантации оплодотворенной яйцеклетки другой женщины ( перенос эмбрионов ).
- Streak гонады с Y-хромосомы , содержащие клетки имеют высокую вероятность развития рака, особенно gonadoblastoma . Streak гонады обычно удаляются в течение года или около этого диагноза , так как рак может начаться в раннем детстве.
эпидемиология
Было подсчитано, что частота синдрома Swyer составляет примерно 1 в 100000 человек. Менее чем 100 случаев были зарегистрированы по состоянию на 2018. Есть очень редкие случаи семейного синдрома Swyer.
история
Синдром Swyer был впервые описан Gim Swyer в 1955 году в докладе двух случаев.
Рекомендации
дальнейшее чтение
внешняя ссылка
Органические причины нарушения полового созревания у подростков | #03/15
Часть 1
Половое созревание это процесс, который начинается в эмбриональном периоде и продолжается в период пубертата, подготавливая организм к репродуктивной функции.
Наступление пубертата контролируется всеми эндокринными железами, но ведущую роль играет гипоталамо-гипофизарная система, которая активирует выработку овариальных и тестикулярных гормонов, оказывающих свое биологическое действие практически на все ткани.
Возраст начала пубертата у детей имеет достаточно широкий диапазон. Прежде всего, он зависит от этнической принадлежности, пола, состояния здоровья, генетической предрасположенности, некоторых факторов внешней среды. Так, холодный климат ассоциирован с более поздним началом полового созревания в популяции. Для детей европейской популяции в 95% случаев пубертат наступает в возрастном интервале от 8 до 13 лет у девочек и от 9 до 14 лет у мальчиков. Половое созревание начинается с появления импульсной секреции лютеинизирующего гормона релизинг-гормона (ЛГ-РГ) в гипоталамусе, который стимулирует выработку в гипофизе фолликулостимулирующего гормона (ФСГ) и лютеинизирующего гормона (ЛГ).
Ранние стадии мужского и женского полового созревания очень похожи. Для становления пубертатной фазы характерно ночное пульсирующее увеличение выброса ЛГ-РГ. Ночной пик ЛГ у мальчиков вызывает более высокий подъем уровня тестостерона. У девочек увеличивающаяся амплитуда колебаний ЛГ стимулирует тека-клетки яичников, в которых вырабатывается тестостерон и в меньшей степени прогестерон. В зернистых клетках яичников под воздействием ФСГ, который вызывает усиление активности ароматазы, основная часть тестостерона конвертируется в эстрадиол. Максимальная концентрация эстрадиола наблюдается в дневное время, т. к. для его образования в яичниках нужен более длительный период времени.
Клинически уровень полового созревания оценивают соответственно критериям шкалы Таннера. Задержкой полового развития считается отсутствие или неполное развитие вторичных половых признаков у детей, достигших максимального возраста нормального пубертата. У мальчиков средний возраст проявления первых признаков полового созревания, а именно увеличение объема тестикул (более 4 мл), около 14 лет. В то время как у девочек развитие грудной железы, как признак начала пубертата, происходит в среднем на 1–2 года раньше [1].
Половое и подмышечное оволосение, которое обусловлено надпочениковыми андрогенами, не является признаком созревания и может встречаться у пациентов с выраженными симптомами гипогонадизма.
Задержка начала созревания может быть связана с нарушением в одном из звеньев гипоталамо-гипофизарно-гонадной цепочки. В подавляющем числе позднее начало пубертата встречается у подростков с семейной предрасположенностью к замедлению роста. В ее основе лежит функциональная незрелость нервных механизмов, которые запускают импульсную секрецию ЛГ-РГ. В этом случае препубертатная скорость роста нормальная, но скелетное созревание и подростковый скачок роста происходят позже [2].
Однако у 0,1% детей позднее начало полового созревания связано с органической патологией. Врожденные или приобретенные аномалии центральной нервной системы (ЦНС) и гипоталамо-гипофизарных структур вызывают полное или частичное нарушение способности гипоталамуса секретировать ЛГ-РГ или гипофиза — вырабатывать ЛГ и ФСГ, что приводит к развитию гипогонадотропного гипогонадизма (гипоГ). Дефект гонад, врожденного или приобретенного характера, которые неспособны к выработке достаточного количества половых гормонов, лежит в основе гипергонадотропного гипогонадизма (гиперГ). Соматические заболевания, приводящие к хронической гипоксии или нарушению белкового обмена, также имеют большое значение в развитии гипогонадизма: синдром нарушенного кишечного всасывания, не компенсированные врожденные пороки сердца, хронические заболевания печени и почек, сахарный диабет и др.
Выделяют три основные формы органического гипогонадизма: первичный, вторичный и гипогонадизм, вызванный нечувствительностью рецепторов к гормонам [3].
Первичный гипогонадизм
Первичный, или гипергонадотропный, гипогонадизм встречается при нарушениях синтеза стероидных гормонов половыми железами. По принципу обратной связи дефицит половых гормонов стимулирует выработку в гипофизе ФСГ и ЛГ. Органические формы гипергонадотропного гипогонадизма чаще связаны с врожденными хромосомными и генетическими аномалиями. Редко встречаются формы гиперГ с отсутствием гонад, но с нормальным хромосомным набором.
Процесс гонадной и генитальной дифференцировки происходит в период раннего эмбриогенеза. Развитие половых желез обусловлено набором половых хромосом (ХХ и ХУ), определяющих генетический пол. Они инициируют трансформацию первичной биопотенциальной гонады в тестикул или яичник, что в конечном счете формирует гонадный пол.
Для нормального формирования мужского фенотипа необходима интактная У-хромосома с локализацией на ней гена SRY (sex-determining on Y chromosome), а также нормальная функция Х-хромосомы с наличием на ней гена, ответственного за рецепторы к андрогенам.
Для последующей дифференциации органов по мужскому типу необходима достаточная секреция тестостерона клетками Лейдига и выработка антимюллерова гормона (АМГ) клетками Сертоли, а также наличие в клетках-мишенях фермента a-редуктазы.
Тестостерон требуется для развития вольфовых протоков, что приводит к образованию из них придатка яичка, семявыносящих протоков и семенных пузырьков. Активность АМГ вызывает регресс мюллеровых протоков. Под воздействием интратестикулярного тестостерона количество семенных канальцев увеличивается во внутриутробном периоде. Кроме того, тестостерон необходим для развития предстательной железы, полового члена и мошонки. В этих органах тестостерон превращается в более активный метаболит дигидротестостерон (ДГТ) под действием фермента 5-a-редуктазы. Интратестикулярный тестостерон необходим для поддержания сперматогенеза и препятствует апоптозу зародышевых клеток.
Дефект в любом звене этого механизма ведет к нарушению половой дифференцировки и развитию органов по женскому типу.
Формирование яичника из первичной гонады происходит позже, чем у плода мужского пола. Предшественники ооцитов окружаются веретенообразными клетками, которые в последующем превращаются в гранулезные клетки. Совокупность ооцита и гранулезных клеток образует примордиальный фолликул. В последующем наружный слой веретенообразных клеток видоизменяется в тека-клетки. Гранулезные и тека-клетки составляют основу стероидсекретирующих структур яичника. Гормональная активность ячников в фетальный период минимальна.
При отсутствии андрогенов в период раннего внутриутробного развития вольфовы протоки редуцируются. Из мюллерова канала формируются маточные трубы, матка и верхняя треть влагалища.
Характер и степень повреждения гонад и генитальной дифференцировки зависит от времени внутриутробного повреждения. На самом раннем этапе дифференциации пола (2–7 неделя) для эмбрионов любого пола мутация аутосомных генов приводит к дисгенезу гонад у плода.
На 7–10 неделе из первичной биопотенциональной гонады формируется тестикул или яичник, что определяется влиянием генов, находящихся на половых хромосомах. Отсутствие гена SRY у детей с нормальным мужским кариотипом приводит к дисгенезии тестикул. Наличие гена SRY у детей с нормальным женским кариотипом приводит к формированию первичной гонады в тестикул (ХХ-мужчины).
Дифференцировка внутренних и наружных гениталий (фенотипический пол) происходит на 9–14 неделе. У плода мужского пола это зависит от функциональной активности эмбриональных тестикул. В этот период формируются различные варианты ложного мужского и женского гермафродитизма. Дефект секреции АМГ у мальчиков приводит к образованию у них матки и фаллопиевых труб в сочетании с бесплодием. Дефект биосинтеза тестостерона и дигидротестостерона — к феминному или бисексуальному строению наружных гениталий при нормально развитых тестикулах. У девочек экстрагонадная секреция тестостерона (ВДКН) вызывает андрогенизацию наружных гениталий при нормально сформированных яичниках. Повышенная экспрессия АМГ приводит к агенезии матки и влагалища (синдром Рокитанского).
Итак, мутация генов, контролирующих процессы половой дифференцировки, в сочетании с мутациями аутосомных генов способствует формированию различных клинических синдромов [4].
Врожденные формы
Синдром Кляйнфельтера
Это генетическое заболевание, характеризующееся дополнительной женской половой Х-хромосомой (47ХХY). Реже встречаются мозаичные формы (46ХY/47XXY). Частота данного синдрома колеблется в пределах 1 на 500–700 новорожденных мальчиков. Патология возникает в результате нарушения расхождения хромосом на ранних этапах формирования яйцеклеток и сперматозоидов. Дополнительная Х-хромосома не влияет на формирование тестикул и наружных гениталий по мужскому типу, но сперматогенез нарушен [5].
Юноши с синдромом Кляйнфельтера, как правило, выше своих сверстников, так как линейный рост продолжается дольше. Формируются евнухоидные пропорции тела: длинные конечности, высокая талия, бедра относительно шире пояса верхних конечностей, избыточное отложение жира в области сосков, живота, у гребешков подвздошных костей; мышцы дряблые, слабые, голос высокий, детский. Характерна пубертатная смешанная гинекомастия. Тестикулы небольших размеров, плотные на ощупь.
Дефицит андрогенов приводит к замедленному или неполноценному половому созреванию, а в дальнешем к бесплодию. Степень вирилизации больных варьирует, но в большинстве случаев отмечается оволосение лобка по женскому типу, а также недостаточный рост волос на лице. Возможна патология урогенитального тракта: крипторхизм, гипоспадия и микропенис.
У части больных выявляются признаки костной дисплазии: «готическое» небо, искривление нижних конечностей, деформация грудины. Со стороны сердечно-сосудистой системы обнаруживают разнообразные врожденные пороки сердца.
По сравнению со здоровыми такие подростки в будущем имеют повышенный риск рака молочной железы, системных заболеваний (красная волчанка и др.). У этих больных могут быть проблемы с обучением, трудности контакта с людьми, но индивидуальные черты личности различаются.
Специфического лечения нет. Проводится пожизненная заместительная гормональная терапия (ЗГТ) препаратами тестостерона, начиная с 13–14 лет.
Синдром Шерешевского–Тернера
Синдром Шерешевского–Тернера (СШТ) представляет собой хромосомную мутацию, которая влияет на рост и развитие девочек. Частота возникновения приблизительно 1 на 2500 новорожденных, но значительная часть приходится на неразвивающиеся беременности и мертворождение. Хромосомная аномалия определяется отсутствием одной Х-хромосомы (моносомия 45ХО) или ее дефектом (45ХО/46ХХ,45ХО/XY)
Наиболее общая черта СШТ — низкорослость, которая очевидна уже в 5-летнем возрасте. До этого возраста темпы роста относительно приемлемы. В патогенензе низкорослости имеют значение генетические нарушения, связанные с делецией гена SHOX. Низкий рост у таких пациенток сочетается с костными аномалиями: девиацией локтевых суставов, искривлением костей голеней, изменением костей позвоночника (короткая шея), укороченем IV и V пальцев рук, пятый палец нередко искривлен, недоразвитие лицевого скелета, «готическое» небо. Характерна остеопения в пубертатном возрасте.
Структурные аномалии Х-хромосомы приводят к двухсторонней дисгенезии гонад. При наличии только одной Х-хромосомы овоциты яичника подвергаются дегенерации еще до рождения. Половой инфантилизм встречается у 95–98% пациентов с СШТ. Ранняя потеря функции яичников при СШТ сопровождается высокой гипоталамо-гипофизарной реакцией. Первое время яичники развиваются нормально, но яйцеклетки (ооциты), как правило, преждевременно редуцируются и большая часть овариальной ткани дегенерирует еще до рождения. Строма яичников фиброзируется, и они превращаются в «strеak»-гонады [6]. В пубертатный период уровень ФСГ резко повышается. Однако у большинства девочек не начинается половое созревание без гормонального лечения, и они не могут забеременеть. Вторичное оволосение — лобковое и подмышечное — развивается спонтанно к 12 годам у всех девочек с СШТ под влиянием надпочечниковых андрогенов. Лишь небольшой процент девочек сохраняют нормальную функцию яичников в юношеском возрасте. Также высокий риск заболеваний щитовидной железы (хронический аутоиммунный тиреоидит, гипотиреоз, гипертиреоз) и развития сахарного диабета 2-го типа.
Около 30% девочек с СШТ имеют дополнительную кожную складку на шее (крыловидная шея), низкий уровень роста волос на задней поверхности шеи, отечность рук и ног (лимфедема), пороки почек (подковообразная, гипоплазия, удвоение мочевыводящих путей). От трети до половины пациенток рождаются с пороком сердца (коарктация аорты, дефект аортального клапана, стеноз легочной артерии и др.). Осложнения, связанные с этими пороками, потенциально опасны для жизни. Высокое артериальное давление распространено у женщин с СШТ. Это может быть связано с коарктацией аорты или заболеванием почек, хотя часто причину найти не удается.
У девочек с СШТ часто развивается хронический средний отит, особенно в раннем возрасте, что в конечном итоге может привести к снижению слуха. Такие дети должны регулярно наблюдаться ЛОР-врачом.
Большинство девушек и женщин с СШТ имеют нормальный интеллект. Но возможны задержки моторного развития и поведенческие проблемы.
Лечение включает в себя несколько направлений. Инъекции гормона роста в раннем детстве позволяют увеличить конечный рост на несколько сантиметров. Терапию эстрогенами рекомендуется назначать в возрасте 15 лет, когда костный возраст достигает максимума. Эстроген-гестагенные препараты применяются в период, когда нужно поддержать фертильность.
Синдром Нунан
Встречается с частотой 1 на 10 000–25 000 всех новорожденных. Фенотипически дети схожи с синдромом Шерешевского–Тернера, но в отличие от последнего имеют нормальный набор хромосом (46XX или 46XY). Заболевание встречается у мальчиков и у девочек [6].
Дети с синдромом Нунан имеют выраженные симптомы дисэмбриогенеза: широко расставленные бледно-голубые или сине-зеленые глаза, низко посаженные уши, высокое арочное небо, небольшую нижнюю челюсть (микрогнатия). У многих детей короткая шея с избытком кожной ткани и низкая линия роста волос на задней части шеи. Около 50–70% низкорослые. При рождении они, как правило, имеют нормальную длину тела и вес, но со временем рост замедляется (низкий уровень соматотропного гормона (СТГ)). Специфическая деформация грудной клетки по типу «куриная грудь» (pectus carinatum) или «воронкообразная грудная клетка» (pectus excavatus) сочетается со сколиозом.
Большинство пациентов имеют порок сердца — наиболее часто стеноз легочной артерии, гипертрофическую кардиомиопатию. Нарушение в свертывающей системе крови проявляются чрезмерной кровоточивостью: длительными кровотечениями после травм или операций. У женщин отмечают меноррагии и родовые кровотечения.
У мальчиков часто встречается задержка полового развития. У большинства из них выявляется крипторхизм и гипоплазия тестикул, фертильность нарушена. Девочки с синдромом Нунан обычно не имеют отставаний в половом развитии и нарушений фертильности.
Большинство детей интеллектуально развиваются нормально, однако встречаются больные с выраженным снижением мнестико-интеллектуальных функций.
Синдром Свайера при 46ХY (чистая дисгенезия)
Для больных с этим синдромом характерен женский фенотип — наружные и внутренние половые органы (матка и фаллопиевые трубы). В пубертатном возрасте отмечается недостаточное развитие вторичных половых признаков, первичная аменорея, гипергонадотропный гипогонадизм. Вместо половых желез у них имеются скопления неразвитой ткани (стрековые гонады), неспособные секретировать тестостерон и АМГ. Кариотип определяется как 46ХY. Эти аномальные половые железы часто малигнизируются, поэтому должны быть хирургически удалены в раннем детстве. Синдром встречается с частотой 1 на 30 000 новорожденных. Гонадный дисгенез вызывается мутацией гена SRY [7].
Пациенты с синдромом Свайера обычно выглядят как женщины и идентифицируют себя соответственно. ЗГТ начинают в подростковом возрасте, чтобы вызвать менструацию и развитие женских вторичных половых признаков, таких как развитие груди и оволосение. ЗГТ также помогает предотвратить остеопороз. Женщины с этим синдромом не имеют собственных яйцеклеток, но могут выносить беременность с помощью донорского эмбриона. В связи с риском перерождения стрековых гонад в злокачественную опухоль рекомендуется их удалять.
Сертолиноклеточный синдром (Дель Кастильо)
Синдром изолированной аплазии сперматогенного эпителия сравнительно редкое заболевание, характеризуется азооспермией при нормальном уровне тестостерона и ЛГ, но повышенном уровне ФСГ. Больные в физическом и половом развитии мало отличаются. Ведущим симптомом заболевания является бесплодие, кариотип нормальный (46XY), в очень редких случаях может отмечаться легкая гипоплазия яичек. При биопсии обнаруживается умеренное уменьшение извитых канальцев, отсутствие предшественников сперматозоидов и зародышевых клеток. Семенные канальцы выстланы только клетками Сертоли. Гистологическая картина напоминает ткань допубертатного тестикула. Функция клеток Лейдига не нарушена. Уровень тестостерона нормальный.
Приобретенные формы
Гипергонадотропный гипогонадизм развивается у больных в результате хирургической операции (опухоль, острый некроз тестикул), воспаления (при вирусном, туберкулезном паротите), телегамма- и химиотерапии. Длительное наблюдение за детьми с различными эндокринопатиями позволило выявить гонадную недостаточность у 42% у них.
Вторичный (центральный, гипогонадотропный) гипогонадизм
ГипоГ формируется в случае нарушения способности секреции гипоталамусом ЛГ-РГ или аденогипофизом — ЛГ и ФСГ. Результатом дефицита гонадотропинов является резкое снижение биосинтеза половых стероидов в тестикулах или в яичниках. Причиной гипоГ может быть врожденная и приобретенная патология гипоталамуса и гипофиза. В подавляющем числе случаев эта форма гипогонадизма диагностируется у юношей. У девушек это проявляется, прежде всего, первичной аменореей, которая может иметь множество других причин, в том числе при anorexia nervosa, травмах и опухолях ЦНС.
В норме ЛГ-РГ секретируется нейронами гипоталамуса. Выделение нейронами этого гормона носит импульсный характер. Последнее является необходимым условием для нормального функционирования гонадотропных клеток гипофиза. ЛГ в мужском организме стимулирует синтез андрогенов (С-19-стероидов), а ФСГ регулирует сперматогенез в тестикулах. В женском организме ЛГ также поддерживает биосинтез андрогенов, ФСГ стимулирует ароматизацию (ферментативная реакция), превращая андрогены в эстрогены. Синтез гормонов осуществляется под контролем различных генов, мутация которых может сопровождаться гипогонадизмом [8].
Клинически гипоГ проявляется симптомами дефицита андрогенов и отсутствием или задержкой полового созревания.
Врожденные формы
Большое количество пациентов с данной патологией свидетельствует о еще не известных мутациях в генах. Исследования в этом направлении привели к открытию нескольких генов, которые необходимы для развития и правильного функционирования ГЛ-РГ-зависимых нейронов гипоталамуса, дисфункция которых может привести к гипоГ без выпадения обоняния и с выпадением, как при синдроме Кальмана.
У отдельных мутаций разная пенетрантность и экспрессивность. Есть два биологических пути, которые могут привести к гипоГ. Первый берет начало в эмбриологии. ЛГ-РГ-секретирующие нейроны мигрируют из обонятельной луковицы к гипоталамусу во время эмбриогенеза. Мутации, которые мешают этой миграции, приводят к гипогонадизму с выпадением обоняния, т. е. к синдрому Кальмана. Второй путь предполагает наличие мутаций, которые вызывают аномальную низкую активность ЛГ-РГ-секретирующих нейронов, прошедших нормальный путь миграции. Соответственно, развивается гипогонадизм без нарушений со стороны обоняния. Заболеваемость врожденным гипоГ встречается с частотой 1–10:100 000 живорожденных, две трети случаев приходится на синдром Кальмана, остальные это идиопатические формы.
Синдром Кальмана
Характеризуется задержкой или отсутствием полового созревания в сочетании с нарушением обоняния [8]. По разным оценкам встречается у 1 на 10 000–86 000 новорожденных, чаще у мальчиков. Исследователи выделяют четыре формы синдрома Кальмана (4-го типа), которые отличаются своей генетической природой. Наиболее распространенной формой является синдром Кальмана 1-го типа. Гипогонадизм при этом синдроме связан с дефицитом ЛГ-РГ. Дефект секреции гонадолиберина приводит к отсутствию или выраженному дефициту ЛГ.
У новорожденных часто выявляют микропенис и крипторхизм. В период полового созревания у юношей не развиваются вторичные половые признаки, такие как рост волос на лице и теле, изменение тембра голоса. Девушки не менструируют, молочные железы формируются плохо или не развиваются вовсе. Больные высокого роста с евнухоидными пропорциями тела.
При синдроме Кальмана обоняние либо уменьшается (гипосмия) или полностью отсутствует (аносмия). Этот специфический признак отличает синдром Кальмана от других форм гипоГ, которые не влияют на обоняние. Нужно учитывать, что часто люди не подозревают о дефекте обоняния, пока это не обнаружится специальными тестами. Обонятельные нейроны, так и нейроны, секретирующие ЛГ-РГ, в период эмбриогенеза совместно формируются, а затем мигрируют в разные отделы мозга. При этом синдроме формирование обоих нейронов происходит нормально, а миграция нарушена.
Клинические симптомы синдрома варьируются даже среди одной семьи: односторонняя почечная агенезия, «заячья губа» и «волчья пасть», «готическое» небо, хаотичные движения глазных яблок, потеря слуха и аномалии развития зубов. У некоторых пациентов диагностируют бимануальную синкенезию, при которой движения одной стороны зеркально повторяются другой. Бимануальная синкенезия сильно затрудняет выполнение задач. требующих действий только одной рукой (игра на музыкальном инструменте).
Лечение заключается в длительном введении гонадотропинов. При резкой гипоплазии яичек наряду с хориогонином применяют препараты тестостерона. В дальнейшем возможно лечение только ХГЧ.
Синдром Паскуалини (синдром плодовитых евнухов)
Редкое заболевание, связанное с врожденной изолированной недостаточностью ЛГ и тестостерона. Содержание ФСГ в пределах нормы, сперматогенез активный. Некоторые авторы выявили у этой группы больных частичный дефицит ЛГ-РГ. Этого количества гормона достаточно, чтобы местно стимулировать клетки Лейдига для выработки тестостерона, необходимого для сперматогенеза и увеличения тестикул.
В пубертатном возрасте такие больные высокорослы, имеют евнухоидные пропорции тела, скудное оволосение на лице, подмышечных впадинах, лобке. Тестикулы нормальных размеров. Сперматогенез не нарушен [9, 10].
Септооптическая дисплазия (мутация гена HESX1, синдром Morsier)
Мутация гена HESX1 ассоциирована с септооптической дисплазией, синдромом врожденного гипопитуитаризма, гипоплазией зрительного нерва и агенезом срединных структур головного мозга. Ген HESX1 кодирует белок, который играет важную роль в раннем развитии мозга [5, 11]. Описано четыре мутации этого гена. Заболевание встречается с частотой 1 на 10 000 новорожденных. Признаки и симптомы данного синдрома варьируют, но существуют три характерные черты: гипоплазия зрительного нерва, нарушение формирование структур вдоль средней линии мозга и гипоплазия гипофиза.
Гипоплазия зрительного нерва вызывает нарушение зрения в одном или обоих глазах. Клинические симптомы в виде косоглазия, нистагма, снижения остроты зрения, а при исследовании глазного дна — атрофии зрительного нерва выявляются уже на первом году жизни. Вследствие задержки визуального развития у младенцев, которые кажутся слепыми, зрение может улучшиться в течение первых нескольких месяцев жизни. У детей с односторонней или ассиметричной двусторонней гипоплазией зрительного нерва снижение зрения стойкое и необратимое.
Характерной чертой дисплазии является неправильное развитие срединных структур мозга (мозолистого тела), разделяющих правое и левое полушарие. Следствием аномалии развития мозга является снижение интеллекта.
Недоразвитие гипофиза приводит к дефициту тропных гормонов, необходимых для нормального роста, полового развития и других важных функций. С возрастом нарастает отставание в физическом развитии и низкорослость. В пубертатном периоде развивается гипоГ. В тяжелых случаях пангипопитуитаризма, когда гипофиз не производит никаких гормонов, в крови наблюдается постоянная гипогликемия.
Симптомы септико-оптической дисплазии могут существенно различаться. Некоторые исследователи предполагают, что они проявляются группой связанных синдромов, а не каким-то одним. Около трети людей с диагнозом дисплазии сочетают все три группы симптомов, у большинства две главные особенности. В редких случаях синдром Morsier ассоциирован с повторными эпилептическими припадками, задержкой развития и нарушением координации.
Дети с пангипопитуитаризмом должны получать соответствующую ЗГТ.
Генетические дефекты формирования аденогипофиза
Более десятка генных мутаций могут вызвать гипоплазию гипофиза, у большинства из них не наблюдается дисплазии зрительных нервов (KAL, GNRHR, PIT1, PROP1). Наиболее изученными являются гены PIT1 и PROP [12]. Мутации этих генов вызывает дефицит тиреотропного гормона (ТТГ), пролактина (ПРЛ), соматотропного гормона (СТГ), ЛГ и ФСГ, который может быть тотальный или парциальный. В детском возрасте чаще встречаются симптомы, связанные с дефицитом СТГ и ТТГ. В период созревания диагностируется отставание в половом развитии.
Мутация гена DAX1, ассоциированная с врожденной гипоплазией надпочечников и гипогонадотропным гипогонадизмом
Мутации в гене DAX1 вызывает Х-хромосомную врожденную надпочечниковую гипоплазию и гипопогонадотропный гипогонадизм. Ген DАX1 кодирует белок, который комплексируется с ядерным ДНК в гипоталамусе и гипофизе [13]. Роль DAX1 и других неопределенных аутосомно-рецессивных генов в развитии коры надпочечников пока не совсем ясна. DAX1 представляется необходимым для окончательной дифференцировки коры надпочечников у взрослых, но не у плода, поскольку последняя сохраняется у пациентов с делецией этого гена. Врожденная гипоплазия надпочечников встречается редко. Клинический опыт показывает, что эта болезнь встречается не так часто, как врожденная гиперплазия коры надпочечников вследствие дефицита 21-гидроксилазы (заболеваемость примерно 1 на 10 000–15 000 новорожденных по всему миру).
Врожденная гипоплазия коры надпочечников это потенциально смертельное заболевание при отсутствии ранней диагностики и лечения. Х-сцепленная врожденная гипоплазия надпочечников связана с дефицитом глицеролкиназы и мышечной дистрофией Дюшена. Дефицит глицеролкиназы приводит к повышению концентрации триглицеридов в крови.
Заболевание встречается только у мальчиков и связано с измененной X-хромосомой.
У заболевших детей после рождения развиваются симптомы первичной глюкокортикоидной и минералокортикоидной надпочечниковой недостаточности и им ошибочно ставят диагноз врожденной дисфункции коры надпочечников. У некоторых больных этот период может пройти относительно благополучно и проявляется в дальнейшем признаками хронической надпочечниковой недостаточности, а в период полового созревания задержкой пубертата [14]. ГипоГ сочетается с первичным дефектом сперматогенеза. У некоторых пациентов была диагностирована нейросенсорная глухота. Возможна внутриутробная задержка физического развития и аномалии наружных половых органов.
Изучение родословной больного позволяет заподозрить гипоплазию коры надпочеников. Похожие симптомы можно отметить у родственников мужского пола (братьев, дядей), но решающую роль в диагностике играет молекулярно-генетический анализ.
Продолжение статьи читайте в следующем номере.
Литература
- Susan Y. Euling, Sherry G. Selevan et al. Role of Environmental Factors in the Timing of Puberty // pediatrics.aappublications.org 2007/.
- Смирнов В. В., Маказан Н. В. Функциональная задержка полового развития: причины, диагностика, лечение // Лечащий Врач. 2012, № 1, с. 30–34.
- Blondell R. D., Foster M. B. et al. Disorders of Puberty // American Family Physician. July. 1999, Vol 60, № 1.
- Reproductive system // Genetics Home Reference. 2013. ghr.nlm.nih.gov.
- Дедов И. И., Семичева Т. В., Петеркова В. А. Половое развитие детей: норма и патология. М., 2002, 232 c.
- Дедов И. И., Петеркова В. А., Семичева Т. В., Волеводз Н. Н. Синдром Шерешевского–Тернера (патогенез, клиника, диагностика, лечение). М.: Pharmacia, 2002. 48 с.
- Шабалов Н. П. Диагностика и лечение эндокринных заболеваний у детей и подростков. М.: МЕДпресс, 2009.
- Migeona C. J.,. Wisniewski A. B. Sexual Differentiation: From Genes to Gender // Hormone Research. 1998; 50: 245–251.
- Hypogonadism in childhood By Mayo Clinic staff. 2011. www.mayoclinic.com.
- Dandona P., Rosenberg M. T. A practical guide to male hypogonadism in the primary care setting // PMC. 2010.
- Коптева А. В., Дзенис И. Г., Бахарев В. А. Генетические нарушения гипоталамо-гипофизарной регуляции репродуктивной системы (обзор литературы) // Проблемы репродукции. 2000, № 3.
- Marino M., Moriondo V. et al. Central Hypogonadotropic Hypogonadism Genetic Complexity of a Complex Disease // PMC. 2014.
- Balsamo A., Antelli A. et al. A new DAX1 gene mutation associated with congenital adrenal hypoplasia and hypogonadotropic hypogonadism // PMC. 2005.
- Nimkarn S., Maria I. New. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia // PMC. 2013.
В. В. Смирнов1, доктор медицинских наук, профессор
А. А. Накула
ГБОУ ВПО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва
1 Контактная информация: [email protected]
Купить номер с этой статьей в pdf
Синдром Суайра. Зачем нужны мужчины
Синдром Суайра
Человеческий эмбрион с кариотипом XY начинает становиться мужчиной только после шестой недели своего развития при условии достаточного количества мужских гормонов. Если вдруг по каким-либо причинам в Y-хромосоме зачатого «мальчукового» эмбриона обнаружится дефект и тестостерон не будет вырабатываться в достаточном количестве или этого гормона в организме матери будет меньше 0,24 нмоль/л, у зародыша могут развиться женские половые органы. По фенотипу (внешним признакам) это будет женщина, а по генотипу (набору генов и хромосом) – мужчина. Именно так проявляется так называемый синдром Суайра.
Встречается это заболевание у одной из 100 000 женщин. Считается, что, несмотря на разовость мутации, синдром Суайра – рецессивный сцепленный признак, то есть он может передаться ребенку по наследству даже от внешне здоровых родителей, если в Х-хромосоме обоих присутствует поврежденный ген. У больных этим синдромом диагностируются недоразвитие наружных половых органов, снижение полового влечения и неизлечимое бесплодие. Нередко синдром Суайра выражается в ненормально высоком для женщин росте или, напротив, в карликовости, у больных отсутствует растительность в подмышечных впадинах и лобковой области, исследования УЗИ показывают отсутствие яичников.
Данный текст является ознакомительным фрагментом.Читать книгу целиком
Поделитесь на страничкеСледующая глава >
Что такое синдром Свайера – ladymady.ru
Синдром персистирующих мюллеровых протоков характеризуется наличием полностью развитых производных мюллеровых протоков у нормально маскулинизированного в остальном генетического мужчины (46.XY). Всего описано менее 200 случаев синдрома. Отсутствие редукции мюллеровых протоков приводит к развитию внутренних женских половых органов у пораженных мужчин при наличии в той или иной степени развитых производных вольфо-вых протоков. Развитие гениталий по промежуточному типу встречается при этом синдроме нечасто. Возможен одно- или двусторонний крипторхизм.
Классификация
В зависимости от уровня поражения гипоталамо-гипофизарной системы, различают следующие формы заболевания:
— гипергонадотропный (первичный) гипогонадизм;
— гипогонадотропный (вторичный) гипогонадизм;
— нормогонадотропный гипогонадизм;
— гипогонадизм, обусловленный резистентностью органов-мишеней.
В зависимости от времени возникновения:
Синдром истинного агонадизма
Синдром рудиментарных яичек
Дисгенез гонад, или синдром Свайера
Агенез клеток Лейдига
Синдром 46ХХ у мужчин, или синдром де ля Шапеля
Синдром ХХY
II. Нарушение развития протоков
У пациенток с чистой формой дисгенезии гонад, или синдромом Свайера, при резко выраженном половом инфантилизмеотсутствуют соматические аномалии развития. Кариотип у больных чаще всего 46, ХХ, 46, XY. Наблюдаемые семейныеслучаи чистой формы дисгенезии гонад вызывают необходимость более тщательного анализа генеалогического деревапациентов. Содержание полового хроматина у большинства больных снижено, но возможно и нормальное его количество(при кариотипе 46, ХХ). Больные с Yхромосомой в кариотипе имеют целый ряд клинических и лечебнодиагностическихособенностей. Помимо задержки полового развития возможна вирилизация наружных половых органов при нормальномполовом оволосении у пациенток с женским типом строения внутренних половых органов и расположением дисгенетичныхгонад в полости малого таза.
ДИАГНОСТИКА
АНАМНЕЗ
Выясняют наличие стигм наследственных и врождённых синдромов и особенностей полового созревания обоих родителейи ближайших родственников (I и II степени родства). Семейный анамнез следует выяснять в процессе беседы только сродственниками пациентки, желательно с матерью. Оценивают особенности внутриутробного развития, течение периодановорождённости, темпы роста и психосоматического развития, выясняют условия жизни и особенности питания девочки смомента рождения, данные о физических, психологических и эмоциональных нагрузках, уточняют возраст и характеропераций, течение и лечение заболеваний, перенесённых по годам жизни, а также семейный анамнез. Позднее менархе уматери и других ближайших родственниц, запоздалое и замедленное половое оволосение и развитие наружных половыхорганов у отца отмечают у большинства девочек с семейной формой ЗПС. У больных с синдромом Кальманна уточняютналичие в семье родственников со сниженным обонянием либо с полной аносмией.
Матери девочек с дисгенезией гонад нередко указывают на воздействие во время беременности физических и химическихвредностей, высокую или частую лучевую нагрузку (рентгеновское, сверхвысокочастотное, лазерное и ультразвуковоеизлучение), обменные и гормональные нарушения, интоксикации на фоне приёма эмбриотоксичных препаратов инаркотических веществ, острые инфекционные заболевания, особенно вирусной природы. До пубертатного возрастаразвитие ребёнка с XY дисгенезией гонад не отличается от сверстников. В пубертатном возрасте, несмотря насвоевременное половое оволосение, развитие молочных желёз отсутствует, менархе не возникает.
ФИЗИКАЛЬНОЕ ИССЛЕДОВАНИЕ
Проводят общий осмотр, измеряют рост и массу тела, фиксируют особенности распределения и степень развитияподкожножировой ткани. Рост и массу тела сопоставляют с регионарными возрастными нормативами. Отмечают признакинаследственных синдромов, рубцы после перенесённых операций, в том числе на черепе. Оценку стадии половогосозревания девочки проводят с учётом степени развития молочных желёз и полового (лобкового) оволосения (критерииТаннера 1969 года с современными поправками).
При осмотре наружных половых органов наряду с оценкой линии роста волос на лобке оценивают форму и размерыклитора, больших и малых половых губ, особенности гимена и наружного отверстия уретры. Обращают внимание наокраску кожи половых губ, цвет слизистой оболочки преддверия влагалища, характер выделений из половых путей.
Осмотр стенок влагалища и шейки матки (кольпоскопию) следует проводить, используя специальные тубусы или детскиезеркала разных размеров с освещением. Для уменьшения диагностических ошибок ректоабдоминальное исследованиецелесообразно проводить после очистительной клизмы, которую назначают больной накануне осмотра.
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
В пубертатном возрасте у больных не появляются вторичные половые признаки: отсутствует оволосение в подмышечных и лобковой областях, неразвиты молочные железы, большие и малые половые губы, матка и влагалище, атрофичная слизистая половых органов. Характерным для синдрома Шерешевского—Тернера является мозаицизм хромосом: X0/XY, ХО/ХХХ, ХО/ХХ и др. При синдроме Шерешевского—Тернера отмечаются существенные гормональные нарушения: высокие уровни ФСГ (в 10—20 раз выше нормы), ЛГ (в 3—4 раза выше нормы), различные аномальные уровни выделения 17-КС (пики повышение — снижение).
Матка и трубы в виде рудиментарных структур, рядом с которыми определяются белесоватые соединительнотканные тяжи (рудиментарные гонады). Описываются различные варианты патологического строения гонад: обычная волокнистая ткань; соединительнотканная строма с интерстициальными клетками, дисгенетические гонады с участками корковой зоны и первичными дегенеративными фолликулами, а также с семенными канальцами; рудиментарные мужские гонады.
«Смешанная» форма дисгенезии гонад — одна из форм интерсексуализма (гермофродитизма), называется также асимметричной. Выделена в нозологическую форму nTSohval в 1905 г.Характеризуется неопределенным фенотипом, с преобладанием в одних случаях женского, в других мужского. При женском фенотипе имеют место интерсексуальное строение половых органов и гипертрофия клитора. При мужском фенотипе в брюшной полости определяется матка с трубами.
«Медицинская генетика и геном человека» - Наука о наследственности и изменчивости. Генетический полиморфизм. Словарь генома. Исследования генома. Statistics. Кариотипирование. Минимальное число генов человека. Муковисцидоз. Ядро. Гены и хромосомы. Мутантные гены. Этиология наследственных болезней. Этапы реализации генетической информации. Cимволы.
Синдром обратного развития половых желез
Сюда входит группа болезней, общим признаком которых является регресс до того сформировавшихся семенных желез. Существует три степени недоразвития яичек: полное отсутствие половых желез, глубокое недоразвитие яичек и отсутствие одного или обоих яичек.
При полном отсутствии половых желез наружные половые органы устроены по женскому типу. Внутренние половые органы недоразвиты, но имеют вид мужских. Болезнь может сочетаться с пороками других органов, неправильным развитием лица ребенка.
При глубоком недоразвитии яичек они значительно уменьшены в размерах, находятся в мошонке либо опускаются не до конца и расположены выше. Понятно, что и функция таких недоразвитых яичек будет в значительной мере снижена. Наружные половые органы сформированы либо как женские, либо как мужские, но размеры полового члена при этом очень резко уменьшены. Все внутренние половые органы являются мужскими.
Отсутствие яичек всегда наблюдается на фоне нормально сформированных внутренних и наружных мужских половых органов.
При всех вышеописанных формах заболевания половое созревание наступает поздно, протекает медленно, функция половых органов резко снижена. Пол ребенка признается мужским, он в дальнейшем неплохо адаптируется в обществе при условии проведения терапии препаратами гормонов и исправлении грубых нарушений хирургическим путем.
Спасибо вам большое! Вы действительно меня успокоили... В какой-то из тем вычитала, что Вам на данный момент не нравится клиника им. Отта, почему? Я записалась на приём к врачу, т.к. восполение половых губ не проходит. Может ли это быть причиной инфекции? Ещё раз, спасибо!
При данной форме дисгенезии гонад имеет место различные нарушения в системе половых хромосом, степень нарушения дифференцировки половой железы может быть различной. Полная утрата или инактивация генов У – или Х-хромомосмы, определяющих пол, приводит к формированию гонадальных тяжей, которые часто представлены недифференцированной соединительной тканью. Гонады обычно лишены герминативного эпителия и не способны к росту и созреванию. Иногда в ткани тяжей обнаруживаются элементы яичка или яичника в зависимости от хромосомной конструкции.
- Синдром нечувствительности к андрогенам. Причины и диагностика
- Гипоплазия и агенезия клеток Лейдига. Причины и диагностика
- Дефекты синтеза андрогенов. Причины и диагностика
- Синдром персистирующих мюллеровых протоков. Дисгенезия гонад
- Истинный гермафродитизм. Причины гениталий по промежуточному типу
- Обследование при развитии гениталий по промежуточному типу. Диагностика гермафродитизма
- Консультация врача при аномалиях половой системы. Смена пола
- Детские гинекологические жалобы. Сбор анамнеза
- Конфиденциальность в детской гинекологии. Опросник HEADS
- Гинекологический осмотр детей. Профилактические осмотры
Ответы Mail.ru: что такое синдром свайера?
Это когда человек не умеет гуглить
Почти то же что "синдром дауна" не умеющего пользоваться поиском !
Синдром Свайера – это врожденное заболевание, которое возникает вследствие сбоя в хромосомах. Больной внешне выглядит как девушка (женский фенотип), но при исследовании полового хроматина определяются X и Y хромосомы, то есть мужской кариотип. Данную патологию также часто называют инверсией пола. Внешне невозможно отличить больную девушку от здоровой. Люди, страдающие синдромом Свайера, имеют женское телосложение, нормальный рост и сформированные внешние половые органы. Зачастую у них физиологически правильно развита матка и фаллопиевы трубы, но иногда бывает их гипоплазия. Яичники сильно видоизменены, они не могут выполнять свои функции. При обследовании на их месте обнаруживаются тяжи из соединительной ткани, могут присутствовать железистые включения. Такие люди полностью стерильны, то есть у них нет фолликулов с яйцеклетками, поэтому забеременеть природным путем они не могут. Вот еще посмотрите <a rel="nofollow" href="http://spinet.ru/conference/forum62.html" target="_blank">http://spinet.ru/conference/forum62.html</a>.
Синдром Лермуайе - причины, симптомы, диагностика и лечение
Синдром Лермуайе – это редкая патология внутреннего уха, обусловленная спазмом артерий лабиринта, характеризующаяся слуховыми и вестибулярными нарушениями. Проявляется двухфазными пароксизмами. В первой фазе присутствует шум в ушах, значительное снижение слуха, во второй слух восстанавливается на фоне приступа головокружения. Диагностируется на основании клинической картины после исключения других заболеваний со сходной симптоматикой. Назначается симптоматическое лечение диуретиками, антигистаминными препаратами и бетагистином. При отсутствии эффекта выполняются хирургические операции.
Общие сведения
Синдром (пароксизм) Лермуайе (лабиринтный ангиоспазм) является редким патологическим состоянием. Впервые был описан французским врачом-оториноларингологом Марселем Лермуайе в 1919 году. Большинство авторов представляют синдром Лермуайе как атипичную форму болезни Меньера, некоторые считают его отдельной нозологией. Дебют заболевания приходится на возраст 30-40 лет, в отличие от болезни Меньера, первые признаки которой выявляются у пожилых людей. У детей подобные состояния встречаются крайне редко. Чаще наблюдается односторонняя симптоматика. В медицинской литературе описаны случаи патологии с единственным, больше не повторявшимся пароксизмом. Иногда попеременно поражается то правое, то левое ухо.
Синдром Лермуайе
Причины
Непосредственные причины, вызывающие синдром Лермуайе, не установлены. Существуют различные предположения относительно этиологии данного заболевания. Считается, что болезнь развивается из-за ангиоспазма, возникающего на фоне аллергической или аутоимунной реакции, приступа мигрени или выброса катехоламинов при психоэмоциональном стрессе. Пароксизмы Лермуайе также могут быть спровоцированы нейропатией VIII черепно-мозгового нерва, обусловленной патологией шейного отдела позвоночника, сосудистыми аномалиями.
Патогенез
Пусковым механизмом пароксизма Лермуайе служит спазм одной из ветвей внутренней слуховой артерии, ведущий к острой гипоксии лабиринта. Нарушается метаболизм. В тканях внутреннего уха накапливаются недоокисленные продукты обмена. Увеличивается сосудистая проницаемость, ткани набухают. Повышается давление жидкости в системе лабиринта – развивается эндолимфатический гидропс. Происходит раздражение рецепторов внутреннего уха. Появляются кохлеовестибулярные нарушения, которые купируются самостоятельно после восстановления кровотока.
Симптомы
Синдром Лермуайе имеет пароксизмальное течение. Сам пароксизм можно разбить на две фазы. В первом периоде наблюдаются кохлеарные симптомы, для второго характерны вестибулярные дисфункции с одновременным улучшением или полным восстановлением слуха. Болезнь манифестирует внезапно среди кажущегося полного здоровья. Некоторые пациенты связывают начало заболевания с недавно перенесённым психоэмоциональным стрессом.
Синдром Лермуайе дебютирует появлением высокотонального тиннитуса (шума в больном ухе). Пациенты описывают его как «писк» или «звон». Интенсивность шума колеблется от небольшой до мучительной, невыносимой. Тиннитус сопровождается ощущением заложенности уха. Больной перестаёт различать высокие звуки. Наблюдается значительное одностороннее снижение слуха вплоть до полной глухоты. Длительность первой фазы составляет от нескольких дней до 2-3 недель. Кохлеарные симптомы постепенно нарастают, достигая своего пика перед приступом головокружения.
Вторая фаза пароксизма короткая, продолжается от 20-30 минут до нескольких часов. На данном этапе синдром Лермуайе проявляется интенсивным системным головокружением. Присутствуют выраженные вестибуловегетативные симптомы – тошнота, рвота, обильное потоотделение. Иногда приступ заканчивается потерей сознания. Патогномоничным признаком лабиринтного ангиоспазма является существенное (вплоть до полного восстановления) улучшение слуха, наступающее на высоте головокружения.
Осложнения
Течение заболевания обычно благоприятное, симптомы обычно полностью купируются после эпизода головокружения. Иногда синдром Лермуайе сопровождается частыми эпизодами ангиоспазма с поражением одного или обоих ушей. Такое течение болезни существенно ухудшает качество жизни пациентов, нередко приводит к возникновению вторичной нейросенсорной тугоухости. Возможны травмы из-за падений в фазе вестибулярных расстройств.
Диагностика
Диагностический поиск при подозрении на синдром Лермуайе осуществляют специалисты в сфере оториноларингологии совместно с неврологами и отоневрологами. Основанием для подтверждения диагноза служат двухфазное течение пароксизма болезни, характерные клинические симптомы. Диагноз устанавливается только после исключения иной патологии, сопровождающейся кохлеарной и вестибулярной дисфункцией. Болезнь Меньера отличается от данного синдрома снижением слуха на низкие тона, головокружение предшествует кохлеарным расстройствам.
Синдром Лермуайе необходимо дифференцировать с невриномой VIII черепного нерва, очаговыми поражениями головного мозга, локализованными в задней черепной ямке и височной области. Шейный вестибулярный синдром, другие лабиринтопатии и лабиринтиты часто дают похожую клиническую картину. В целях дифференциальной диагностики выполняются следующие исследования:
- Визуализационные методики. Одним из визуализационных методов исследования является рентгенография. На рентгенограмме височных костей может обнаруживаться характерное для невриномы преддверно-улиткового нерва расширение внутреннего слухового прохода. Рентгенография позвоночника даёт возможность исключить деформацию шейного отдела. С помощью КТ и МРТ головного мозга, внутреннего уха выявляются заболевания ЦНС, патология ЛОР органов.
- Измерение остроты слуха. При аудиометрии, импедансометрии определяется тугоухость, устанавливается наличие флюктуации слуха (колебание его остроты). Электрокохлеография позволяет подтвердить наличие признаков эндолимфатического гидропса. Органическое поражение ЦНС, вертебробазилярная недостаточность обычно не приводят к нарушению слуха. При вестибулярном нейроните, лабиринтитах не наблюдается значительных колебаний его остроты.
- Эндоскопические методы. Отомикроскопия и эндоскопия уха используются для исключения наружных и средних отитов. С помощью данных исследований можно обнаружить признаки перилимфатической фистулы тимпаногенного происхождения. При синдроме Лермуайе патологические изменения уха отсутствуют.
- Исследование вестибулярных функций. Осмотр пациента выявляет признаки нарушения равновесия. Наблюдается шаткость походки, неустойчивость в позе Ромберга. При исследовании нистагма с помощью очков Френзеля, методом электронистагмографии и видеоокулографии определяется горизонтальный или горизонтально-ротаторный нистагм. Направление быстрого компонента нистагма зависит от фазы течения болезни.
Лечение синдрома Лермуайе
Этиотропные и патогенетические лечебные мероприятия не разработаны. Симптоматическая консервативная терапия направлена на устранение тяжело переносимых острых симптомов болезни – головокружения с тошнотой и рвотой, интенсивного тиннитуса. В большинстве случаев патология подлежит консервативному лечению, не является показанием для хирургического вмешательства. Операции осуществляются при неэффективности консервативной терапии.
Консервативная терапия
Для снижения давления эндолимфы в лабиринте пациенту рекомендуется ограничить употребление жидкости, поваренной соли. Следует полностью отказаться от алкоголя. В целях устранения дисфункции вестибулярного аппарата показана лечебная физкультура. При подборе фармакологического лечения к каждому больному требуется индивидуальный ступенчатый подход. У некоторых пациентов синдром Лермуайе поддаётся терапии антигистаминными препаратами, кортикостероидными гормонами. Широко используется бетагистин, седативные средства.
Для улучшения обменных процессов назначаются нейропротекторы, витамины А, Е, С, группы В. Острый пароксизм обычно купируется в амбулаторных условиях. Пациенту рекомендуется соблюдать постельный режим. При выраженной симптоматике – интенсивном головокружении, многократной рвоте – больной нуждается в стационарном лечении. Для устранения симптомов пароксизма применяются блокаторы м-холинорецепторов, диуретики и нейролептики.
Хирургическое лечение
Хирургические вмешательства осуществляются при неконтролируемых частых мучительных пароксизмах, возникающих на фоне консервативного лечения. Снизить эндолимфатическое давление помогает установка вентиляционной трубки в барабанную перепонку. Другой часто выполняемой операцией является декомпрессия эндолимфатического мешка. С целью уменьшения шума в ушах применяются деструктивные методы – разрушение лабиринта с помощью химического (абляция спиртом, гентамицином), физического (ультразвуковая, крио- или лазеродеструкция) или механического способов.
Прогноз и профилактика
Синдром Лермуайе отличается от других лабиринтопатий и классического варианта болезни Меньера благоприятным течением. Описаны случаи с единственным в жизни пациента эпизодом заболевания. Данная патология крайне редко осложняется нейросенсорной тугоухостью. Прогноз для восстановления вестибулярных функций хороший. Меры, предотвращающие возникновение вестибулокохлеарных нарушений, на сегодняшний день не разработаны.