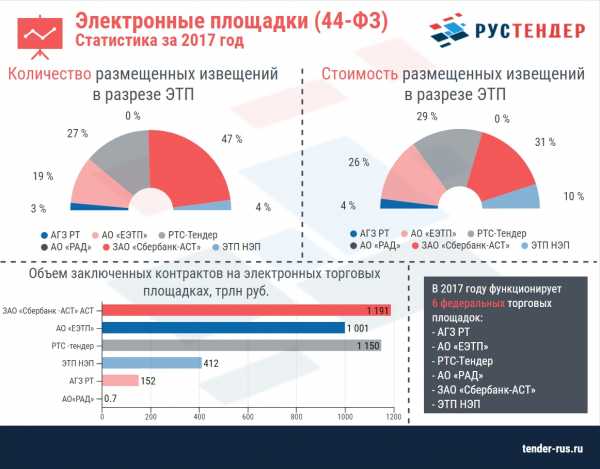
Синдром вискотта олдрича у детей что это такое фото
Синдром Вискотта — Олдрича — Википедия
Синдром Вискотта — Олдрича (СВО, англ. Wiskott–Aldrich syndrome — WAS) — редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении (с уменьшением количества и размеров тромбоцитов), иммунодефицита, и кровавого поноса (обусловленного тромбоцитопенией). Синоним — синдром экземы-тромбоцитопении-иммунодефицита в соответствии с оригинальным описанием Олдрича (англ. Aldrich), сделанным в 1954 году.
В 1937 году немецкий педиатр Вискотт (нем. Alfred Wiskott) описал трёх братьев с низким количеством тромбоцитов (тромбоцитопенией), кровавым поносом, экземой и рецидивирующими инфекциями уха, в то время как их четыре сестры были здоровы. Вискотт охарактеризовал её как болезнь Верльгофа [3].
В 1954 году американский педиатр Олдрич (англ. Robert Anderson Aldrich) на семье американцев голландского происхождения показал, что этот синдром наследуется как Х-сцепленный рецессивный признак[4].
Сочетание синдрома Вискотта — Олдрича с Х-сцепленной тромбоцитопенией встречается у 4—10 из 1 млн родившихся живыми. Географический фактор не имеет значения.
Болеют почти исключительно лица мужского пола. Случаи заболевания лиц женского пола, хоть и описаны в литературе, но очень редки и связаны с подавлением активности одной из X-хромосом[5][6].
Женщины в подавляющем большинстве случаев выступают лишь в роли гетерозиготных носителей и сами почти никогда не болеют, однако для женщин с врождённой тромбоцитопенией оценка гена WASP может оказаться важным диагностическим исследованием[7].
Ген WAS, мутации в котором приводят к развитию данного заболевания, локализован на коротком плече Х-хромосомы. Возникновение мутации в гене, ответственном за синтез белка WASp, приводит к появлению дефектной формы белка или к полному его отсутствию, что влечёт за собой развитие нарушений иммунитета и гемостаза.
Известно более 350 мутаций в гене WAS, которые приводят к иммунодефициту и тромбоцитопении[8][9].
Тип мутации гена WAS существенно коррелирует со степенью тяжести заболевания: те, которые привели к производству усечённого белка, сопровождаются значительно более тяжёлыми симптомами, чем те, которые кодируют нормальную длину белка WASp. Аутоиммунные заболевания и злокачественные новообразования могут произойти при обоих типах мутаций, но пациенты с усечённым геном WAS подвергаются повышенному риску [10].
Наследование[править | править код]
Как любое другое Х-сцепленное рецессивное заболевание, Синдром Вискотта — Олдрича наследуется следующим образом.
Женщина-носитель при каждой беременности имеют 25%-ную вероятность родить здоровую дочь-носителя (как она сама), 25%-ную вероятность родить здоровую дочь без носительства мутантного гена, 25%-ную вероятность родить больного сына и 25%-ную вероятность родить здорового сына. Это также означает, что каждая дочь имеет 50 % шанс стать носителем, а каждый сын имеет 50 % вероятность наличия заболевания[8].
Примерно у 1/3 пациентов с вновь диагностированным СВО причиной заболевания является новая мутация, произошедшая во время оплодотворения яйцеклетки, то есть не унаследована от матери[11].
При WAS снижается количество или вовсе не продуцируется белок WAS (WAS-protein, WASP). Уникальные функции WASP до конца ещё не изучены, однако установлено, что он играет ключевую роль в полимеризации белка актина и формировании цитоскелета. В последнем процессе (в формировании цитоскелета — микрофибрилл, филоподий, фагоцитарных вакуолей и т.д.) принимает непосредственное участие белок миозин, концентрация которого значительно снижена в тромбоцитах больных с WAS. WASP экспрессируется только в клетках гемопоэтической системы. WASP имеет исключительное значение для передачи сигнала от поверхностных рецепторов клетки к цитоскелету, что динамически регулируется им. Это приводит к дефектам формирования всех клеточных структур, образование которых зависит от цитоскелетной реорганизации актиновых филаментов и в результате нарушения многих функций клеток, что в норме экспрессируют WASP, а именно лейкоцитов и тромбоцитов.
Полноценная функция актинового цитоскелета крайне важна уже на стадии продукции тромбоцитов мегакариоцитами в костном мозге, а также для реализации их адгезивных, агрегационных и других функций. Тромбоцитопения и уменьшения размера тромбоцитов (диаметром менее 1,5 мкм при норме 2,3 мкм) является постоянным симптомом при данной патологии.
Основными причинами этого являются:[править | править код]
Число тромбоцитов колеблется от 30 × 109 / л до 140 × 109 / л, но периодически снижается до 10-30 ×109 / л. В пунктате костного мозга определяют отсутствие мегакариоцитов или наличие их дегенеративных форм. В большинстве случаев геморрагический синдром усиливается на фоне инфекций. У больного может также развиться хроническая постгеморрагическая анемия и увеличение селезёнки (спленомегалия).
Иммунная система пациентов с WAS производит очень мало B- и T- лимфоцитов, которые необходимы для защиты организма от инфицирования. Существенно нарушается хемотаксис WASP-дефицитных лейкоцитов, снижается пролиферативный ответ лимфоцитов, нарушается формирование иммунных синапсов Т-лимфоцитов, значительно ослабляется цитолитическая активность натуральных киллеров, также ухудшается IgG-опосредованный фагоцитоз и соответственно, нарушается презентация антигенов. Вот почему пациенты с WAS страдают на повторные бактериальные, грибковые и вирусные инфекции.[12]
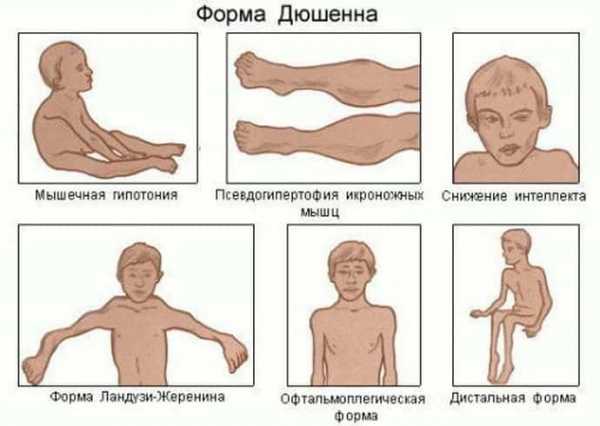
Синдром Вискотта — Олдрича поражает мальчиков и проявляется следующими симптомами: атопическим дерматитом, геморрагическим синдромом (снижением количества тромбоцитов, гемоглобина, эритроцитов) и комбинированным дефицитом В- и Т-лимфоцитов, который ведёт к повторяющимся инфекционным процессам (частые ОРЗ, бронхолёгочные инфекции, инфекции ЛОР-органов, кожи, слизистых, мочевыводящих путей и ЖКТ). Поскольку число тромбоцитов снижено, первым симптомом может быть кровоточивость, например кровавая диарея. Изменение уровня сывороточных иммуноглобулинов — низкий IgM, высокий IgA, очень высокий IgE, уровень IgG может быть нормальным, пониженным или повышенным [13]. Дефицит В- и Т- лимфоцитов делает детей восприимчивыми к заболеваниям, вызванным бактериями, вирусами и грибами. Распространены инфекционные поражения дыхательных путей. У детей, доживших до 10-летнего возраста, развивается по крайней мере одно аутоиммунное заболевание; до трети пациентов страдают онкологическими заболеваниями, в основном это лимфомы и лейкозы[14].
В 1994 году[15] синдром Вискотта — Олдрича был связан с мутациями в гене на коротком плече X-хромосомы, продукт этого гена был назван англ. Wiskott–Aldrich syndrome protein (белок синдрома Вискотта — Олдрича). Позже было открыто, что Х-сцепленная тромбоцитопения объясняется мутацией гена WAS. Кроме того, редкое заболевание — Х-сцепленная нейтропения — связана с особой мутацией гена WAS.
WASp кодируется геном WAS, который содержит 502 аминокислотных остатка; в основном выражен в клетках гемопоэза (эти клетки вырабатываются красным костным мозгом и затем развиваются в клетки крови). Точная функция белка WASp ещё не известна, но были предложены сигнальная трансдукция и поддержание цитоскелета (при СВО нарушен синтеза белка, необходимого для полимеризации актина в клетках и формирования цитоскелета).
Иммунодефицит обусловлен снижением выработки антител, также повреждаются Т-лимфоциты (то есть иммунодефицит является комбинированным). Это ведёт к повышенной восприимчивости к инфекционным заболеваниям, в особенности глаз и ЛОР-органов.
Существует более лёгкая форма СВО, названная Х-сцепленная тромбоцитопения (X-linked trombocytopenia — XLT). При обследовании мальчиков с этим заболеванием, не имеющих других признаков СВО, были выявлены более 60 мутаций в гене WAS, аналогичные таковым при классическом синдроме Вискотта — Олдрича. По неизвестным[8] причинам XLT отличается более мягким течением без выраженных признаков иммунодефицита[16].[17][18]
Радикальное[править | править код]
Поскольку у больных синдромом Вискотта — Олдрича снижено количество тромбоцитов, а тромбоциты разрушаются в селезёнке, спленэктомия часто помогает уменьшить проявления геморрагического синдрома, но не излечивает другие нарушения, характерные для СВО.
Эффективна трансплантация костного мозга, однако трансплантация осложняется трудностью в выборе донора, возможной гипофункцией трансплантата, риском отторжения (реакция «трансплантат против хозяина») и частыми посттрансплантационными осложнениями в виде вирусной, бактериальной и грибковой инфекции.
Консервативное[править | править код]
Экзему сдерживают при помощи местных или системных стероидов, а также общим тщательным уходом за кожей[19].
При массивных кровотечениях показано переливание крови (пациентам с глубоким падением Т-клеточного иммунитета показаны только облучённые Ле-фильтрованные препараты крови).
Возможно проведение заместительной терапии эритроцитарной массой и тромбоконцентратом.
Для лечения, а часто и для профилактики заболеваний бактериальной этиологии применяют Антибиотики (цефалоспорины, аминогликозиды, полусинтетические пенициллины, сульфаниламиды).
Многим пациентам в профилактических целях показан длительный приём противогрибковых препаратов.
Для увеличения количества тромбоцитов применяют агонисты тромбопоэтиновых рецепторов[19]: ромиплостим и элтромбопаг.
Целесообразно переливание внутривенных иммуноглобулинов.
Генотерапия[править | править код]
Начаты исследования коррекции синдрома Вискотта — Олдрича методом генотерапии, используя лентивирус[20][21]. Доказана принципиальная возможность успешной генотерапии гемопоэтических стволовых клеток у пациентов с синдромом Вискотта — Олдрича[22]. В настоящее время исследователи продолжают развивать оптимизированные векторы генотерапии[10][23]. В июле 2013 итальянский San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) сообщил, что на трёх детях с синдромом Вискотта — Олдрича показано существенное улучшение спустя 20-30 месяцев после применения генетически модифицированного лентивируса[24]. Последние испытания генной терапии продемонстрировали обнадёживающие результаты[25] для пациентов, которым невозможно подобрать подходящего донора стволовых клеток (костного мозга)[19].
Эта стратегия может как обеспечить клиническую пользу, так и привести к расширению и злокачественной трансформации гемопоэтических клонов с переносными векторными вставками вблизи онкогенов, что увеличит риск лейкеми[24].
Прогноз очень зависит от того, какая именно мутация в гене WAS вызвала заболевание[8][9][10]. В общем случае без применения трансплантации костного мозга прогноз неблагоприятный. С применением трансплантации — обнадёживающий.
В связи с тем, что антибиотикотерапия, переливание компонентов крови и трансплантация стволовых клеток костного мозга стали более доступны, медиана выживаемости увеличилась с 8 месяцев у пациентов, родившихся до 1935 года, до 6 лет у пациентов, родившихся после 1964 года. По современным данным медиана продолжительности жизни составляет от 8 до 11 лет. В случае, если не применялась трансплантация стволовых клеток костного мозга, молодые пациенты имеют больше шансов умереть от кровотечения, дети чаще умирают от инфекций, а дети и подростки чаще всего умирают от злокачественных новообразований. Лимфомы встречаются у 26 % больных в возрасте от 20 лет и старше. Общий риск злокачественных онкологических заболеваний более чем в 100 раз превышает средний по популяции, а с возрастом увеличивается ещё сильнее[26].
Трансплантация стволовых клеток костного мозга, если удалось преодолеть реакцию «трансплантат против хозяина», приводит к нормализации показателей крови и делает прогноз благоприятным. Выживаемость после применения стволовых клеток продолжает улучшаться.
Прогноз тем благоприятнее, чем меньше времени прошло между установлением диагноза и трансплантацией гемопоэтических стволовых клеток, если её выполнять в возрасте пациента до 5-6 лет и до возникновения значительных осложнений.
Несмотря на то, что СВО остаётся тяжёлым заболеванием, при котором возможны угрожающие жизни осложнения, многие больные мужского пола доживают до подросткового или даже взрослого возраста, ведут продуктивную жизнь и имеют собственные семьи. Самым старшим из больных, получивших трансплантацию костного мозга, сейчас больше тридцати лет, и они кажутся выздоровевшими без развития злокачественных опухолей или аутоиммунных заболеваний[11].
Режим[править | править код]
В связи с высокой возможностью заражения на время обострения необходимо помещать больных с синдромом Вискотта — Олдрича в бокс. По тем же причинам больным с синдромом Вискотта — Олдрича противопоказано нахождение в детском коллективе. Также противопоказаны прививки живыми вакцинами (существует вероятность того, что вакцинный штамм вируса может вызвать заболевание) и препаратами, содержащими полисахаридные антигены (непонятно, выработаются ли нужные антитела).
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Wiskott A. Familiarer angeborener Morbus Werlhofii. Monastsschr // Monastsschr. Kinderleil. Kd. — 1937. — Vol. 68. — P.212-216.
- ↑ Aldrich R. A., Steinberg A. G., Campbell D. C. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. (англ.) // Pediatrics. — 1954. — Vol. 13, no. 2. — P. 133—139. — PMID 13133561.
- ↑ Wengler G., Gorlin J. B., Williamson J. M., Rosen F. S., Bing D. H. Nonrandom inactivation of the X chromosome in early lineage hematopoietic cells in carriers of Wiskott-Aldrich syndrome. (англ.) // Blood. — 1995. — Vol. 85, no. 9. — P. 2471—2477. — PMID 7537115.
- ↑ Davis B. R., Yan Q., Bui J. H., Felix K., Moratto D., Muul L. M., Prokopishyn N. L., Blaese R. M., Candotti F. Somatic mosaicism in the Wiskott-Aldrich syndrome: molecular and functional characterization of genotypic revertants. (англ.) // Clinical immunology (Orlando, Fla.). — 2010. — Vol. 135, no. 1. — P. 72—83. — doi:10.1016/j.clim.2009.12.011. — PMID 20123155.
- ↑ Takimoto T., Takada H., Ishimura M., Kirino M., Hata K., Ohara O., Morio T., Hara T. Wiskott–Aldrich syndrome in a girl caused by heterozygous WASP mutation and extremely skewed X-chromosome inactivation: a novel association with maternal uniparental isodisomy 6. (англ.) // Neonatology : journal. — 2015. — 24 January (vol. 107, no. 3). — P. 185—190. — doi:10.1159/000370059. — PMID 25633059.
- ↑ 1 2 3 4 A service of the U.S. National Library of Medicine: Genetics Home Reference
- ↑ 1 2 WAS — Wiskott–Aldrich syndrome (eczema-thrombocytopenia) Архивировано 1 февраля 2014 года., Resource of Asian Primary Immunodeficiency Diseases (RAPID)
- ↑ 1 2 3 Jin, Y.; Mazza, C.; Christie, J.; Giliani, S.; Fiorini, M.; Mella, P.; Gandellini, F.; Stewart, D. et al. (2004). «Mutations of the Wiskott–Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation». Blood 104 (13): 4010. doi: 10.1182/blood-2003-05-1592 . PMID 15284122 .
- ↑ 1 2 Синдром Вискотта — Олдрича, International Patient Organisation for Primary Immunodeficiencies (неопр.) (недоступная ссылка). Дата обращения 3 августа 2013. Архивировано 18 февраля 2014 года.
- ↑ Синдром Вискотта — Олдрича — первичный комбинированный иммунодефицит (неопр.).
- ↑ Sande, Merle A.; Wilson, Walter P. (2001). Current diagnosis & treatment in infectious diseases. New York: Lange Medical Books/McGraw-Hill. p. 361. ISBN 0-8385-1494-4.
- ↑ Donald A Dibbern Jr, MD; Chief Editor: Michael A Kaliner, MD. Wiskott–Aldrich Syndrome. Medscape.
- ↑ Derry, J. M. J., Ochs, H. D., Francke, U. Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell 78: 635—644, 1994. Note: Erratum: Cell 79: following 922, 1994. doi: 10.1016/0092-8674(94)90528-2 . PMID 8069912.
- ↑ Rosen F. S., Cooper M. D., Wedgwood R. J. The primary immunodeficiencies. (2). (англ.) // The New England journal of medicine. — 1984. — Vol. 311, no. 5. — P. 300—310. — doi:10.1056/NEJM198408023110506. — PMID 6429535.
- ↑ Zhu Q., Watanabe C., Liu T., Hollenbaugh D., Blaese R. M., Kanner S. B., Aruffo A., Ochs H. D. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype. (англ.) // Blood. — 1997. — Vol. 90, no. 7. — P. 2680—2689. — PMID 9326235.
- ↑ Sullivan K. E. Recent advances in our understanding of Wiskott-Aldrich syndrome. (англ.) // Current opinion in hematology. — 1999. — Vol. 6, no. 1. — P. 8—14. — PMID 9915548.
- ↑ 1 2 3 Синдром Вискотта — Олдрича. Резюме (рус.) (pdf). orpha.net (2013). Дата обращения 7 января 2017.
- ↑ Galy, A.; Roncarolo, M. G.; Thrasher, A. J. (2008). «Development of lentiviral gene therapy for Wiskott Aldrich syndrome» . Expert Opinion on Biological Therapy 8 (2): 181—190. doi: 10.1517/14712598.8.2.181 . PMC 2789278. PMID 18194074
- ↑ Frecha, C; M G Toscano , C Costa , M J Saez-Lara , F L Cosset , E Verhoeyen & F Martin (2008). «Improved lentiviral vectors for Wiskott–Aldrich syndrome gene therapy mimic endogenous expression profiles throughout haematopoiesis». Gene Therapy 15 (12): 930-41. doi: 10.1038/gt.2008.20. PMID 18323794
- ↑ Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P. P.; Díez, I. S. A.; Dewey, R. A.; Böhm, M.; Nowrouzi, A. et al. (2010). «Stem-Cell Gene Therapy for the Wiskott–Aldrich Syndrome» . New England Journal of Medicine 363 (20): 1918—1927. doi: 10.1056/NEJMoa1003548. PMC 3064520. PMID 21067383
- ↑ Dewey, R.; Diez, I.; Ballmaier, M.; Filipovich, A.; Greil, J.; Gungor, T.; Happel, C.; Maschan, A. et al. (2006). «Retroviral WASP gene transfer into human hematopoietic stem cells reconstitutes the actin cytoskeleton in myeloid progeny cells differentiated in vitro». Experimental Hematology 34 (9): 1161—1169. doi: 10.1016/j.exphem.2006.04.021 . PMID 16939809
- ↑ 1 2 Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M. P.; Baricordi, C.; Dionisio, F.; Calabria, A. et al. (2013). «Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott–Aldrich Syndrome». Science. doi: 10.1126/science.1233151.
- ↑ Worth A. J., Thrasher A. J. Current and emerging treatment options for Wiskott-Aldrich syndrome. (англ.) // Expert review of clinical immunology. — 2015. — P. 1—18. — doi:10.1586/1744666X.2015.1062366. — PMID 26159751.
- ↑ Perry G. S. 3rd, Spector B. D., Schuman L. M., Mandel J. S., Anderson V. E., McHugh R. B., Hanson M. R., Fahlstrom S. M., Krivit W., Kersey J. H. The Wiskott-Aldrich syndrome in the United States and Canada (1892-1979). (англ.) // The Journal of pediatrics. — 1980. — Vol. 97, no. 1. — P. 72—78. — PMID 7381651.
причины, симптомы, диагностика, лечение, прогноз
Синдром Вискотта-Олдрича (СВО) — наследственная патология, обусловленная дефицитом особого белка WASp, обеспечивающего взаимодействие между клетками крови. Он принимает непосредственное участие в процессе свертывания крови при повреждении кровеносных сосудов, а также в поддержании иммунной защиты организма от патогенных и условно-патогенных микробов. СВО относится к группе первичных иммунодефицитных состояний, обусловленных поражением Т- и В — лимфоцитов, продуцирующих антитела. Дефицит тромбоцитов приводит к повышенной кровоточивости и массивной кровопотере. Заболевание проявляется триадой симптомов — экземой, первичным иммунодефицитом и тромбоцитопенией.
СВО является редкой Х-сцепленной рецессивной патологией, при которой женщины считаются носителями мутантного гена. При этом сами они не болеют, а передают поврежденный ген детям. Это легко объяснить наличием у них здоровой аналогичной хромосомы, которая не позволяет недугу развиться. У сыновей болезнь проявится клинически, а дочери становятся носителями мутантного гена.
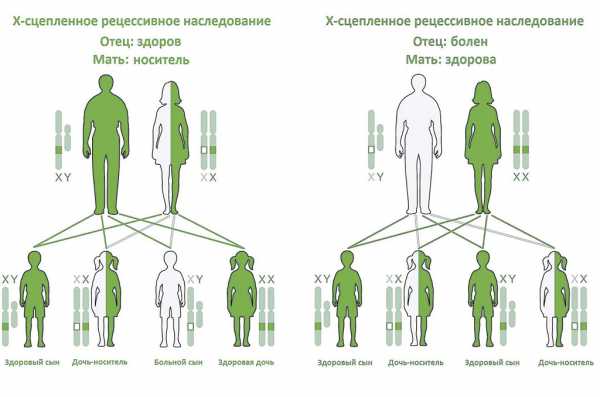
Принципы наследования СВО
Синдром впервые был описан в 1937 году немецким педиатром Вискоттом. Он наблюдал за тремя братьями, у которых имелись проявления тромбоцитопении, рецидивирующих инфекций уха, экземы. В 1954 году детский врач из Америки Олдрич определили характер наследования заболевания. Спустя много лет был выявлен ген, мутации которого приводят к синдрому. В 50-х и 60-х годах 20-го века синдром вошел в список первичных иммунодефицитов на основании признаков иммунной недостаточности у больных.
Патология иммунной системы развивается в период эмбриогенеза. Врожденный иммунодефицит приводит к развитию тяжелых инфекционных недугов. Он часто сочетается с тромбоцитопенией и экземой на лице, конечностях или всего тела. У больных СВО значительно повышается риск развития злокачественных опухолей и аутоиммунных нарушений. Дети с данной патологией в наибольшей степени подвержены бактериальной, грибковой или вирусной инфекции.
СВО встречается у 4-10 из 1 миллиона родившихся живыми детей. Географический фактор при этом не имеет значения. Страдают патологией исключительно мужчины. Женщины же являются гетерозиготными носителями патологии.
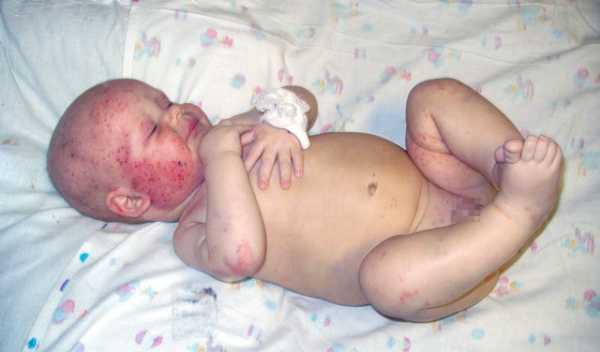
ребенок с синдромом Вискотта-Олдрича
Выделяют три формы СВО:
- Легкая форма – микротромбоцитопения, иммунодефицит, отсутствие экземы, нечастые инфекции, проходящие без осложнений;
- Среднетяжелая и тяжелая формы фактически мало чем отличаются друг от друга и проявляются экземой, плохо поддающимися лечению аутоиммунными, инфекционными и онкологическими заболеваниями.
Этиология
СВО — генетически детермированная патология, в основе которой лежит мутация гена, ответственного за синтез белка, функция которого до конца не известна. Его дефект, недостаток или полное отсутствие приводят к нарушению иммунной защиты и работы свертывающей системы крови. У больных с СВО в результате мутации развивается иммунодефицит и тромбоцитопения.
Иммунодефицит обусловлен неспособностью иммунокомпетентных клеток воспринимать информацию об антигенах и продуцировать антитела. Подобные расстройства связаны с нарушениями белкового обмена. При снижении иммунной защиты организма возникают острые инфекции различной этиологии у людей с СВО.
Поражение белковых молекул, участвующих в процессе тромбоцитообразования, приводит к снижению количества и ухудшению качества этих клеток. В результате возникают частые кровотечения, экземы, язвы на коже. Аномально уменьшенные в размерах тромбоциты перестают выполнять свои функции в полном объеме и разрушаются в селезенке. Это приводит к тромбоцитопении и спленомегалии. У больных часто появляются синяки и петехии, кровотечения из носа и десен, кровавый понос. Причиной плохого самочувствия у таких больных становится анемия.
Основные типы мутации генов при СВО:
- Мутации гена, приводящие к образованию усеченного белка, вызывают СВО с тяжелым течением и ярко выраженной симптоматикой. Когда выработка белка полностью прекращается, возникает «классическая», самая опасная, форма болезни.
- Мутации гена, кодирующего нормальную длину белка, приводят к развитию типичной формы СВО. Выработка некоторого количества измененного белка проявляется среднетяжелым течением синдрома.
Оба типа генетических мутаций могут привести к аутоиммунным заболеваниям и злокачественным новообразованиям у больных.
Мутантный ген, носителем которого является беременная женщина, передается по наследству детям следующими способами:
- в 25% случаев беременности рождается здоровая дочь-носительница,
- в 25% – здоровая дочь,
- в 25% – больной сын,
- в 25% – здоровый сын.
Симптоматика
СВО проявляется симптомами атопического дерматита и геморрагического синдрома. Дефицит В- и Т-лимфоцитов приводит к часто повторяющимся и тяжело протекающим инфекционным процессам. У больных развиваются заболевания ЛОР-органов, бронхов и легких, кожи, почек и пищеварительного тракта.
Чаще всего организм поражают инфекции, вызванные условно-патогенными микроорганизмами — золотистым стафилококком, гемолитическим стрептококком, синегнойной палочкой, некоторыми энтеробактериями, а также патогенными грибками и вирусами. Бороться с оппортунистическими инфекциями очень сложно, поскольку аутофлора, вызывающая воспаление, присутствует на всей поверхности тела человека. У больных часто рецидивирует герпетическая инфекция, проявляющаяся ангинами, стоматитом, поражением половых органов. Активируются вирусы Эпштейна-Барр, цитомегаловирус. Организм не может противостоять бактериальной атаке, поскольку имеется неполноценность белков, подавляющих размножение микробов и уничтожающих их.
Симптомы СВО впервые проявляются на первых месяцах жизни. С возрастом они прогрессируют. У мальчиков с синдромом Вискотта – Олдрича возникают следующие клинические признаки:
- наружные кровотечения при травматическом повреждении, кровоточивость десен, носовые кровотечения;
- внутренние кровотечения — желудочно-кишечное, подкожное, внутрисуставное;
- признаки анемии — головокружение, слабость, упадок сил, тошнота;
- гематурия, кровь в кале, кровавая рвота;
- боль в суставах;
- экзема — поражение кожи аллергической природы с образованием гиперемированных пятен, язв и эрозий на лице, конечностях и ягодицах;
- зудящие шелушащиеся высыпания на коже, напоминающие клиническую картину атопического дерматита и возникающие в межсезонье в ответ на аллергенный агент.
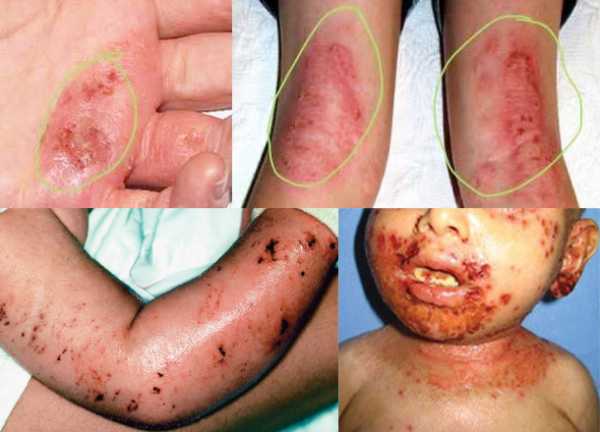
Проявления синдрома Вискотта-Олдрича
На фоне кровопотери снижается общий гемоглобин и возникает гипоксия тканей. Компенсаторно повышается давление и увеличивается нагрузка на миокард и стенки сосудов. Так развивается сердечная недостаточность. Эндотелий сосудов легко повреждается и плохо восстанавливается, приводя к опасным для жизни сосудистым катастрофам.
Тяжесть клинических проявлений может варьироваться от проходящей тромбоцитопении с незначительными геморрагическими признаками до тяжелого заболевания с выраженным симптомами инфекционных и аутоиммунных нарушений. Симптомы СВО ухудшают общее состояние больных и выбивают их из привычного ритма жизни.
Дети, дожившие до 10 лет, обычно страдают хотя бы одним, а чаще несколькими, аутоиммунными заболеваниями – васкулитом, гемолитической анемией, полиартритом. Постоянные васкулиты приводят к гибели пациентов от острой коронарной или мозговой недостаточности. Подавление активности иммунных клеток или снижение их количества приводит к развитию онкозаболеваний — лейкоза или лимфомы. Осложнениями частых инфекционных заболеваний становятся тяжелые пневмонии и сепсис.
Диагностические мероприятия
Синдром Вискотта-Олдрича подозревают у всех мальчиков с кровотечениями и врожденной тромбоцитопенией. Признаки острых инфекций и аутоиммунных расстройств могут отсутствовать или быть слабо выраженными.
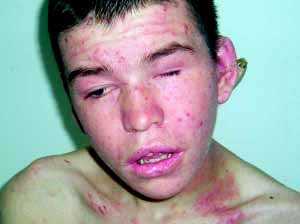
16-летний мальчик с СВО
- Чтобы подтвердить или опровергнуть предполагаемый диагноз, специалисты собирают анализ и выслушивают жалобы больных. Особого внимания заслуживает время возникновения кровотечения, его характер, симптомы инфекционных болезней.
- Поскольку СВО является наследственным заболеванием, очень важен анализ семейного анамнеза. Его выявление у родственников считается важным диагностическим критерием.
- Затем специалисты переходят к общему осмотру больного, во время которого обнаруживают многочисленные гематомы, петехии и экземы.
- Данные гемограммы — тромбоцитопения, анемия.
- Иммунограмма — снижение иммуноглобулинов М, повышение иммуноглобулинов A и E, нормальный уровень иммуноглобулинов G.
- В ходе генетического исследования выявляют мутации в гене, кодирующем синтез белка, ответственного за иммунную защиту организма.
- Больным с СВО показана консультация специалистов в области аллергологии, иммунологии, гематологии, медицинской генетики.
Диагностика СВО является сложной и многоуровневой, определяющей дальнейшее лечение больных.
Лечение
Синдром Вискотта-Олдрича относится к неизлечимым наследственным патологиям. Все лечебные манипуляции носят исключительно симптоматический характер, поскольку невозможно изменить геном человека. Их основная цель — уменьшить выраженность клинических проявлений и облегчить состояние больных, не допустив дальнейшего прогрессирования недуга и развития опасных осложнений.
Консервативная терапия
Терапевтические процедуры позволяют лишь увеличить продолжительность и повысить качество жизни больных.
- Пациентам проводят иммуномодулирующую и иммуносупрессивную терапию: вводят иммуноглобулины и назначают курсовой прием цитостатиков. Иммуносупрессоры подавляют собственные иммунокомпетентные клетки для того, чтобы не отторгался трансплантат.
- Для лечения экземы применяют местные и системные кортикостероиды. Десенсибилизирующие средства и гормональные мази снимают зуд и гиперемию экземных проявлений, а также оказывают противоаллергическое действие.
- При выраженных признаках геморрагического синдрома показано переливание крови, эритроцитарной массы и тромбоконцентрата. Объемные инфузии восполняют недостаточность свертывающей системы крови.
- Препараты железа назначают больным с анемией – «Сорбифер Дурулес», «Феррум лек», «Гемофер».
- Противомикробное лечение показано всем больным с признаками инфекционного поражения внутренних органов. Обычно используют антибиотики из группы цефалоспоринов, макролидов, фторхинолонов, пенициллинов.
- Противовирусные препараты – «Валтрекс», «Ацикловир», «Ингавирин».
- Противогрибковые препараты назначают часто с профилактической целью – «Нистатин», «Флюконазол», «Кетоконазол».
Хирургическое лечение
Трансплантация костного мозга проводится в специализированных клиниках. В настоящее время этот метод является основным и самым эффективным в лечении СВО. Пациентов помещают в стерильный бокс. Подобная изоляция позволяет минимизировать их контакты с патогенными биологическими агентами — бактериями, вирусами, простейшими, грибами. После относительной стабилизации общего состояния больной получает костный мозг донора, который заранее подбирают по гистосовместимости и особым способом подготавливают для пересадки. После успешного завершения реабилитационного периода новая, хорошо прижившаяся ткань начинает полноценно функционировать. Трансплантация костного мозга — эффективная лечебная процедура, нередко осложняющаяся проблемами с поиском донора, риском разрушения пересаженной ткани и частым развитием посттрансплантационных осложнений. Пересаженные стволовые клетки продуцируют достаточное количество здоровых тромбоцитов. Правильно подобранная поддерживающая терапия позволяет иммунной системе пациента «привыкнуть» к чужеродным тканям и не пытаться их ликвидировать. Эта операция отличается высокой стоимостью и отсутствием гарантий.
Спленэктомия помогает уменьшить проявления геморрагического синдрома, но не вылечить его. В селезенке тромбоциты подвергаются массивному разрушению. Удаление органа спасает больных от кровотечений. При этом повышается риск развития септических состояний. После операции у больных повышается число тромбоцитов в крови и увеличивается их размер. У детей, перенесших спленэктомию, намного чаще возникают острые инфекционные заболевания, чем у остальных.
Генотерапия в настоящее время разрабатывает способы, с помощью которых возможно изменение набора генов в пораженных клетках. Для этого необходимо внести в клетку нужный ген, а дефектный удалить, чтобы не дать ему возможности запустить патологический процесс.
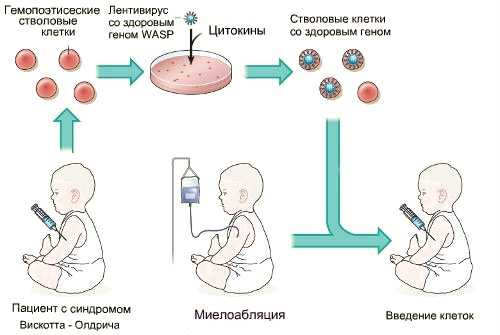
пример современного метода лечения СВО
Чтобы избежать развития острой инфекции, аутоиммунного заболевания или онкопатологии, больным необходимо соблюдать рекомендации врачей:
- не прививаться живыми вакцинами,
- не давать больным детям аспирин и другие препараты из группы НПВС,
- избегать травм, ушибов, порезов,
- маленьким детям носить каски и щитки, защищающие голову и суставы от повреждений,
- укреплять иммунитет,
- ограничивать контакты со сверстниками,
- дважды в сутки проводить влажную уборку с дезинфицирующим средством,
- мыться с использованием специальных гипоаллергенных средств,
- соблюдать гипоаллергенную диету.
Без лечения прогноз СВО неблагоприятный. Смерть больных может наступить от кровопотери или септических осложнений. Периодические курсы поддерживающей терапии увеличивают продолжительность жизни больным и улучшают ее качество. Многие мальчики превращаются в зрелых мужчин, работают, заводят семьи.
В настоящее время пересадка костного мозга — единственно правильный метод лечения, эффективность которого резко падает без здорового гистосовместительства. Все больные должны пожизненно соблюдать рекомендации врачей и принимать назначенные ими медикаменты.
Видео: о детях с синдромом Вискотта-Олдрича
Синдром Вискотта-Олдрича - причины, симптомы, диагностика и лечение
Синдром Вискотта-Олдрича – это иммунодефицитное наследственное заболевание, которое характеризуется экземой, геморрагическим синдромом, высокой вероятностью развития опухолей, инфекционных и аутоиммунных болезней. Ключевыми симптомами являются длительные кровотечения при травмах, частые носовые кровотечения и гематомы, рецидивирующие бактериальные, грибковые и вирусные инфекции, кожная сыпь, провоцирующая зуд и чувство жжения. Диагноз устанавливается на основе клинико-анамнестического обследования, результатов общего анализа крови, исследования иммунитета и биогенетического теста. Лечение включает трансплантацию гемопоэтических клеток и паллиативную терапию.
Общие сведения
Как синоним синдрома Вискотта-Олдрича (СВО) используется термин синдром экземы-тромбоцитопении-иммунодефицита, а также аббревиатура WAS, происходящая от английского «Wiskott-Aldrich syndrome». Заболевание названо по фамилиям американских исследователей. Э. Вискотт в 1937 году описал клинические случаи тромбоцитопении в сочетании с экземой и рецидивирующими инфекциями у мальчиков из одной семьи, в которой все девочки были здоровы. Р. Олдрич в конце 80-х и начале 90-х годов установил, что патология является наследственной и сцепленной с полом, определил расположение гена с дефектом. Распространенность синдрома крайне низкая – 1 случай на 1 млн. новорожденных мальчиков. Отмечены единичные случаи данной болезни у девочек, что связано с недостаточной инактивацией дефектной хромосомы X.
Синдром Вискотта-Олдрича
Причины
СВО – наследственная болезнь. Причиной ее развития является мутация в гене WAS, ответственном за синтез белка иммунных клеток. Ген локализован в X-хромосоме в регионе Хр11.23. В настоящее время выявлено более 300 вариантов мутаций гена WAS, которые приводят к развитию X-сцепленной тромбоцитопении, чаще всего – к микротромбоцитопении и нейтропении. От характера мутации зависит степень проявления симптомов – от снижения уровня тромбоцитов, выявляемого только лабораторным методом, до тяжелой формы синдрома Вискотта-Олдрича с присоединением онкозаболевания крови, аутоиммунной патологии.
Тип наследования синдрома – X-сцепленный рецессивный. У мужчин имеется лишь одна X-хромосома, если она оказывается дефектной, заболевание проявляется. Отец передает мутацию всем дочерям, сыновья получают нормальную хромосому Y. Женщины имеют две X-хромосомы. Дефектный ген, расположенный в одной из них, инактивируется, так как является рецессивным. Синдром не проявляется, но женщины остаются носительницами мутации и с вероятностью 50% могут передать ее детям обоих полов (сыновья болеют, дочери – носительницы).
Патогенез
Генетический дефект проявляется нарушением синтеза белка WASP, который присутствует в клетках системы иммунитета. Он производится гемопоэтическими стволовыми клетками – самыми ранними предшественниками элементов крови, расположенными в красном костном мозге. WASP участвует в перестройке и восстановлении цитоскелета, формировании иммунологических синаптических передач, внутриклеточном транспорте белков. При его отсутствии или недостаточном производстве образуются дефектные тромбоциты, патологически изменяются функции клеток, отвечающих за реакции врожденного и адаптивного иммунитета.
Ухудшается процесс свертывания крови, развивается прогрессирующий иммунодефицит. Степень выраженности клинических проявлений синдрома частично зависит от клеточной концентрации WASP, а его количество определяется локализацией и типом мутации. Симптомы вариативны, у большинства пациентов диагностируется болезнь слабой и умеренной тяжести. Тяжелое течение встречается редко.
Классификация
Единая классификация синдрома Вискотта-Олдрича отсутствует. В клинической практике распространено использование балльной системы, в основе которой лежит утверждение, что у всех больных имеется тромбоцитопения, у большей части – иммунодефицитное состояние, а остальные симптомы могут отсутствовать. С учетом этих параметров выделяют три формы заболевания:
- Легкий СВО. Характеризуется отсутствием экземы или легкой, поддающейся лечению экземой, редкими инфекциями. Оценивается в 1-2 балла.
- Классический среднетяжелый СВО. Проявляется рецидивами инфекционных болезней и экземы, умеренными аутоиммунными реакциями. Соответствует 3-м и 4-м баллам.
- Классический тяжелый СВО. Пациенты страдают от рекуррентных инфекций, тяжелой экземы, аутоиммунных патологий и злокачественных новообразований. Паллиативное лечение малоэффективно. Выраженность синдрома – 5 баллов.
Симптомы
Клинические признаки тромбоцитопении обнаруживаются с рождения. У младенцев на поверхности кожи видна петехиальная сыпь – небольшие красные пятна, образовавшиеся по причине разрыва мелких кровеносных сосудов и медленной сворачиваемости крови. Выявляются экхимозы – подкожные кровоизлияния пурпурного и голубовато-черного диаметром от 3 мм. Стул содержит примеси крови. Наблюдаются частые кровотечения из носа, кровавая рвота. Снижение свертываемости крови отмечается при инвазивных медицинских процедурах, например, после забора крови для анализа.
Экзема обычно проявляется с детства, но может отсутствовать на протяжении всей жизни. У малышей до года она схожа с себорейным или пеленочным дерматитом. Возможно развитие генерализованной формы, при которой поражается кожа всего тела ребенка, или локальной, характеризующейся высыпаниями на конечностях. В старшем возрасте экзема нередко ограничивается областями в локтевых сгибах, вокруг кистей рук, под коленными суставами, в складках кожи шеи. У некоторых больных сыпь отсутствует или проявляется крайне редко и слабо, не требует лечения.
Иммунологические нарушения характерны для большинства пациентов. Снижение защитных функций организма приводит к частым отитам, пневмониям, кандидозам, менингитам, энтероколиту, герпесу и кожным инфекционным патологиям. Развиваются хронические формы инфекций с частыми рецидивами, эффективность стандартной терапии низкая. У взрослых возникают аутоиммунные заболевания – больные подвержены гемолитической анемии, васкулитам, тромбоцитопенической пурпуре, воспалительным поражениям почек и кишечника. При классическом тяжелом СВО у подростков и молодых людей формируются злокачественные новообразования, наиболее частым вариантом является неходжкинская лимфома.
Осложнения
При СВО отмечается высокий риск летального исхода. В 59% случаев причиной смерти становятся инфекционные заболевания. Среди них преобладают тяжелые пневмонии и сепсис (заражение крови). На фоне инфекций усиливается геморрагический синдром, а интенсивное расчесывание зон, пораженных экземой, провоцирует развитие грибковых и бактериальных болезней кожи. Около 21% больных погибают в результате внутренних кровотечений. Жизнеугрожающими являются внутричерепные, желудочно-кишечные и внутрилегочные кровоизлияния. У 12% пациентов причиной ранней гибели становится онкопатология, наиболее распространены лимфомы и лейкозы.
Диагностика
Симптомы тромбоцитопении у детей с синдромом Вискотта-Олдрича проявляются сразу после рождения, но диагноз подтверждается на первом-втором году жизни. Первичная диагностика осуществляется неонатологами и педиатрами при участии генетиков. В ходе дифференциальной диагностики исключается тромбоцитопеническая пурпура, наследственная тромбоцитопения с талассемией и X-сцепленный врожденный дискератоз. Проводится различение с лейкоплакией, атрезией слезных желез, анемией и ненаследственной тромбоцитопенией. Комплексное обследование включает следующие процедуры:
- Опрос. В беседе с родителями выясняется наличие тромбоцитопений и тяжелых рецидивирующих инфекций среди родственников мужского пола, смертей мальчиков в раннем возрасте из-за инфекций, кровоизлияний и рака крови. Жалобы включают кожную сыпь, кровотечения, частые инфекционные болезни.
- Физикальное обследование. При осмотре отмечаются петехии, экхимозы, проявления атопического дерматита, инфекционные очаги на коже. Методом пальпации определяется увеличение размеров лимфоузлов, печени и селезенки (лимфаденопатия, гепатоспленомегалия).
- Лабораторные исследования. Анализы крови являются наиболее информативными методами диагностики. Выполняется ряд тестов:
- Клинический анализ крови. Диагностируется тромбоцитопения с уменьшением размеров тромбоцитов. Этот признак уникален, позволяет дифференцировать СВО с другими видами тромбоцитопений.
- Исследование иммунного статуса. С раннего возраста в иммунограмме обнаруживается лимфоцитопения с наиболее выраженным снижением количества CD8 лимфоцитов. Выявляется нарушение реакций гиперчувствительности замедленного типа, снижение показателя В-лимфоцитов, снижение уровня иммуноглобулинов M и G, повышение концентрации иммуноглобулинов A и E.
- Исследование WASP. Экспрессия белка определяется в клеточных культурах. Результат позволяет с большой вероятностью подтвердить диагноз и составить прогноз заболевания. Полное отсутствие производства WASP отмечается при тяжелых формах синдрома.
- Молекулярно-генетический анализ. Обнаружение мутации гена WAS выполняется методом ПЦР с последующим секвенированием продуктов реакции. Наличие дефектного гена подтверждает предполагаемый диагноз. В отдельных случаях характер мутации учитывается при составлении прогноза.
Лечение синдрома Вискотта-Олдрича
Терапия проводится методом трансплантации гемопоэтических стволовых клеток или костного мозга. Пересадка биоматериала здорового человека позволяет полностью устранить гематологический и иммунологический дефекты. При подборе гистосовместимого донора эффективность такого лечения достигает 84-90%. До внедрения трансплантации ГСК в медицинскую практику большинство пациентов доживали лишь до 6-6,5 лет.
Дополнительно назначается паллиативная терапия, которая представлена внутривенным введением иммуноглобулина, профилактическим применением противовирусных и противомикробных лекарств. При выраженном геморрагическом синдроме может быть проведена спленэктомия – хирургическое удаление селезенки. После процедуры отмечается увеличение количества и размера тромбоцитов. При острых бактериальных инфекциях показано лечение антибиотиками широкого спектра действия.
К новым разработкам в лечении СВО относится генная терапия с использованием лентивирусов. Суть метода заключается в заборе стволовых аутологичных клеток у пациента, последующей коррекции генетического дефекта в этих клетках с применением лентивирусного вектора и введении модифицированного материала в организм. В процессе терапии осуществляется уничтожение клеток дефектной иммунной системы. Проводятся экспериментальные исследования данного метода, получены сообщения о его высокой эффективности.
Прогноз и профилактика
Современные методы лечения позволили значительно увеличить продолжительность и повысить качество жизни больных. При комплексном подходе к лечению прогноз благоприятный. Профилактика синдрома невозможна, поскольку заболевание является наследственным. Для расчета вероятности рождения больного ребенка необходимо медико-генетическое консультирование супружеских пар из группы риска. Для предупреждения осложнений рекомендуется строго соблюдать правила гигиены, исключить контакты с инфекционными больными, избегать травматизации, использовать специальные гипоаллергенные средства для ухода за кожей ребенка, придерживаться диеты, исключающей попадание аллергенов с пищей.
Симптомы синдрома Вискотта Олдрича и его лечение у детей и взрослых + фото

Синдром Вискотта-Олдрича – наследственное заболевание, обусловленное снижением количества (вплоть до отсутствия) одного из белков (WASp), который необходим для взаимодействия между форменными элементами крови.
Заболевание сцеплено с Х-хромосомой, т.е. носителями патологического (рецессивного) гена являются женщины. Вероятность рождения больного мальчика составляет 25%. Женщины являются только носителями синдрома Вискотта-Олдрича, заболевание у них никак не проявляется.
Сущность синдрома Вискотта-Олдрича

Синдром проявляется только у мальчиков, женщины — лишь носители
При синдроме Вискотта-Олдрича наблюдается врождённый иммунодефицит, т.е. патологии иммунной системы развиваются ещё во внутриутробном периоде.
Из-за нарушения белкового обмена клетки иммунной системы не в состоянии нормально передавать и сохранять информацию об антигенах (чужеродные белки) и синтезировать антитела. Это, в свою очередь, приводит к постоянным тяжким грибковым, вирусным и бактериальным инфекциям у людей с синдромом Вискотта-Олдрича.
Бактериальные, вирусные, грибковые заболевания не оставляют пациента с синдромом Вискотта-Олдрича
Белок WASp также важен во время эпизодов кровотечения, поскольку участвует в процессе формирования тромбоцитов. У таких больных наблюдается снижение количества и ухудшение качества этих клеток, что приводит к кровоточивости, экземам, язвам на лице и конечностях. Последний симптом может быть достаточно тяжким – язвочки поражают всю поверхность тела.
Особенности течения соматических заболеваний при диагнозе
Кроме постоянных ОРВИ, кишечных инфекций, дерматитов различной природы и туберкулёза, больные с синдромом Вискотта-Олдрича страдают от собственной условно-патогенной флоры. Это т.н. оппортунистические инфекции ушей, глотки, носа, дыхательных путей и глаз. Бороться с ними крайне непросто, т.к. флора, вызывающая воспалительный процесс есть повсюду, включая поверхности тела самого человека: стафилококки, клостридии, стрептококки и т.п.
Частые сопутствующие заболевания (фото)
- Стоматит
- Герпес
- ЛОР-инфекции
- Туберкулез
Грибок поражает не только кожу, но и слизистые желудочно-кишечного тракта, половые органы, ногти. Могут быть даже грибковые менингиты и менингоэнцефалиты.
У таких больных постоянно рецидивирует герпес, причём может проявляться достаточно обширно, в виде герпетических ангин, стоматита. Это тот случай, когда один человек может последовательно переживать атаки нескольких типов герпетической инфекции. Например, после обострения опоясывающего герпеса начинается генитальный и наоборот.
Активируются также другие вирусы этого же семейства – Эпштейна-Барр, цитомегаловирус и т.п.
Дефицит и плохое качество существующих тромбоцитов приводит к постоянным кровотечениям. Кровоточат дёсны, любые порезы становятся серьёзной проблемой. При небольших воспалительных процессах в кишечнике может появляться кровь в кале.
Алая кровь на поверхности кала – кровотечения из нижних отделов (толстый кишечник) пищеварительного тракта. Чёрная кровь (т.н. мелена) – из верхних.
Больные ходят в синяках из-за постоянных микротравм кожи. По этой же причине часто болят суставы (кровоизлияния в суставную сумку).
На фоне постоянной потери крови снижается общий гемоглобин и ухудшается снабжение тканей кислородом.
В качестве компенсационного механизма может повышаться давление, т.е. увеличивается нагрузка на сердечную мышцу и стенки сосудов. Это, в свою очередь, провоцирует сердечную недостаточность и сосудистые катастрофы, т.к. стенка сосуда легко повреждается и плохо восстанавливается.
Язвы и эрозии на коже (экзема) преимущественно располагаются на ягодицах, лице и конечностях. Этот симптом имеет определённую сезонность, т.к. по природе является аллергическим.
Диагностика синдрома Вискотта-Олдрича
Рано или поздно человеку с синдромом Вискотта-Олдрича понадобится помощь генетика
Оценивается частота различных заболеваний, их тяжесть и характер жалоб. Особенное внимание обращают на заболевания кожи – тяжёлые и с неясной этиологией. Если кроме заболеваний кожи есть кровотечения, например, понос с кровью, врач должен связать эти факты и предпринять диагностические мероприятия для исключения синдрома Вискотта-Олдрича. В пользу этого говорит также наличие синдрома (или склонности к кровотечениям) у родственников больного.
Внимательно осматривается кожа на предмет гематом, экзематозных дефектов.
Диагностика синдрома уже у 3х месячного плода — далеко не редкость в современной медицине
Назначается анализ крови, в которой отмечается снижение количество тромбоцитов. При более детальных исследованиях видно, что размер этих клеток также изменён.
Биохимические показатели указывают на отсутствие WASp белка, а генетическое исследование подтверждает наличие самого заболевания.
Современная пренатальная диагностика позволяет определить наличие синдрома Вискотта-Олдрича у плода уже на 3м месяце беременности. Если семейный анализ указывает на возможность этой генетической аномалии – стоит проверить плод сразу.
Для диагностики состояния могут быть привлечены медицинский генетик, гематолог, иммунолог и аллерголог.
Лечение синдрома методом пересадки костного мозга
Пересадка костного мозга — наиболее безопасный и эффективный метод лечения
Разумеется, перепрошить геном невозможно, поэтому все лечебные мероприятия носят более или менее симптоматический характер.
Наиболее этиопатогенетический (причинно-следственный) метод лечения – пересадка костного мозга.
В этом случае пациенту пересаживают нормальные стволовые клетки, вырабатывающие здоровые тромбоциты (и другие форменные элементы крови) в достаточном количестве.
Процедура пересадки гемопоэтических клеток производится в специальных клиниках, оборудованных асептическими боксами (стерильные палаты).
Пациент вынужден постоянно принимать иммуносупрессоры (препараты, подавляющие собственный иммунитет), профилактические дозы антибиотиков и поддерживающую терапию.
Гипотетически – через некоторое время новый костный мозг приживётся и заменит старый, а иммунитет пациента каким-то образом «привыкнет» к чужеродным тканям и не будет пытаться их ликвидировать.
На практике – донорский костный мозг должен быть идеально совместим с геномом пациента, иначе сразу после отмены иммуносупрессоров он будет атакован иммунокомпетентными клетками и уничтожен.
Второй из возможных негативных исходов – атака хозяина самим костным мозгом, т.н. реакция «трансплантат против хозяина». Новые клетки белой крови, вышедшие из пересаженного мозга, имеют своё «правильно», т.е. начинают атаковать все ткани организма хозяина, считая его чужаком и агрессором (в природе не принято меняться костным мозгом).
Нужно учитывать и тот факт, что данная манипуляция производится далеко не везде и стоимость её может быть крайне высока, при отсутствии гарантий.
Оценка рисков и факторы, влияющие на благоприятную пересадку костного мозга
Самое главное – найти прямого родственника-донора. Если совпадают все необходимые параметры, то костный мозг приживается более чем в 70% таких случаев, т.е. манипуляция имеет смысл.
Пересадка костного мозга от условно-совместимого донора эффективна лишь в трети случаев.
Эту статистику трансплантологи не слишком любят афишировать, поскольку далеко не в каждом случае удаётся найти подходящего родственника. Можно к этому прибавить тот факт, что 70% успешных пересадок костного мозга включают аутотрансплантации, т.е. пересадки собственного костного мозга пациента, т.е. этот показатель для родственников должен быть несколько ниже.
Необходимо понимать, что неудачная трансплантация костного мозга может значительно укоротить продолжительность жизни. Это связано с замещением старого костного мозга новым. В случае аутоиммунной реакции из-за гистологической несовместимости, пациент остаётся вообще без стволовых клеток.
Критерии эффективности пересадки костного мозга при синдроме Вискотта-Олдрича (фото)
- Отсутствие цитамегаловируса
- Молодость пациента
- Родственник-донор
Степень соответствия с донорским костным мозгом оценивается по международной системе HLA.
Вторым критерием является возраст и состояние пациента – чем моложе, тем лучше прогноз.
Следующий критерий – шедевр, основное назначение которого – защитить клинику от тяжб после неудачных трансплантаций. Звучит так: «отсутствие цитомегаловирусной инфекции у реципиента и донора».
Для понимания смысла этого критерия, достаточно знать тот факт, что цитомегаловирус есть приблизительно у 80% взрослого населения нашей страны. Это один из группы герпетических вирусов, тихо (и пожизненно) существующий почти у каждого.
Кроме того, этот вирус наследуется от матери к ребёнку.
Шансы на отсутствие цитомегаловируса у двух родственников без врождённого иммунодефицита всегда менее 4%.
Больной с синдромом Вискотта-Олдрича имеет цитомегаловирус практически всегда, из-за низкой устойчивости к вирусам.
Лечение осложнений
Переливание крови могут периодически проводиться для поддержания состояния пациента
В зависимости от конкретной ситуации назначаются антибиотики, противовирусные и/или противогрибковые препараты. Больным показаны периодические переливания компонентов крови (плазма, эритроцитарная масса), назначение препаратов, увеличивающих свёртываемость и прочие меры профилактики кровотечения.
Для стимуляции костного мозга используются специальные факторы роста, влияющие на тот или иной кровяной росток – тромбопоэтин, эритропоэтин.
Ввиду постоянного иммунодефицита таким больным необходима поддерживающая терапия иммуноглобулинами, интерлейкинами и другими имуннокорректорами.
Экзему лечат при помощи гормональных и антигистаминных препаратов.
Основной лечащий врач – гематолог.
Грамотная стимуляция тромбоцитарного ростка в сочетании с цитокинотерапией позволяет значительно улучшить качество и продолжительность жизни у больных с синдромом Вискотта Олдрича.
Сопутствующие заболевания и исходы
Все инфекции у таких больных протекают тяжёло. Любой бронхит или ОРВИ достаточно легко модифицируется в пневмонию. У таких пациентов сепсис может развиться из-за небольшой царапины или обострения любого хронического инфекционного процесса.
Очень часто формируются аутоиммунные заболевания – быстро прогрессируют ревматизм, ревматоидный артрит, псориаз и многие другие процессы.
Из-за постоянных васкулитов (воспаление стенки кровеносного сосуда) такие пациенты часто погибают от инсультов и инфарктов.
Отсутствие иммунокомпетентных клеток приводит к быстрому появлению и развитию злокачественных опухолей, в т.ч. лейкозов и лимфом.
Профилактика осложнений синдрома Вискотта-Олдрича
Особые условия проживания уберегут людей с синдромом Вискотта-Олдрича от осложнений и сопутствующих проблем
Комплекс мероприятий напоминает подобное у больных ВИЧ и, одновременно, больных гемофилией. Начинается с периода новорожденности и продолжается до конца жизни.
С одной стороны, проводится постоянное укрепление иммунитета, с другой – профилактика травм, ушибов. Необходимо ограничивать контакты со сверстниками, проводить влажную уборку в помещении 2 раза в сутки с применением методов дезинфекции.
Обстановка в помещении должна быть лишена острых углов и прочих травмоопасных элементов. Одежда на ребёнке должна быть прочной. Желательно – вшивать щитки, защищающие суставы от повреждений.
Купание лучше производить с использованием лечебных ванн и специального (банного) мыла. После купания необходимо смазывать кожу увлажняющим кремом.
Пациентам показана диета со сниженным содержанием потенциальных аллергенов. Исключается молоко, шоколад, цитрусовый, все специи, арахис.
Для составления индивидуального меню необходима консультация диетолога.
Детям с синдромом Вискотта-Олдрича ни в коем случае нельзя вводить живые вакцины!

Детям с синдромом Вискотта-Олдрича вводить живые вакцины строго запрещено!
При врождённом иммунодефиците, характерном для этого состояния, даже ослабленные штаммы возбудителей могут быть опасны для ребёнка (собственно, и для взрослого тоже).
Итак, вот что нужно помнить о синдроме Вискотта-Олдрича:
- Женщины являются бессимптомными носителями, диагностировать заболевание можно уже на 3м месяце беременности;
- Пересадка костного мозга является единственным концептуальным методом лечения, однако без здорового гистосовместимого родственника эффективность метода резко падает;
- Соблюдение рекомендаций врача и пожизненный приём медикаментов позволяют существенно увеличить продолжительность жизни людей с синдромом Вискотта-Олдрича.
что это, симптомы, диагностика и варианты лечения
Иммунодефицитные состояния характеризуются нарушением основных функций иммунной системы, снижением защитных сил организма и развитием серьёзных нарушений. Они значительно снижают качество жизни больного и требуют длительного, иногда пожизненного лечения. В число таких нарушений входит синдром Вискотта-Олдрича — редкая болезнь, которая поражает сразу несколько разновидностей клеток иммунной системы, сопровождается кровотечениями и другими тяжёлыми симптомами, а также повышенным риском развития осложнений.
Что такое Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича — наследственная патология, которая относится к первичным иммунодефицитным состояниям. Это врождённое заболевание, сопровождающееся поражением Т-лимфоцитов и В-лимфоцитов — клеток, отвечающих за деятельность иммунной системы и свёртывание крови. В результате они перестают нормально выполнять свои функции или становятся полностью неработоспособными, а способность организма к тромбообразованию и самостоятельной остановке кровотечений снижается. Согласно международной классификации МКБ-10, синдрому Вискотта-Олдрича был присвоен код D82.0.
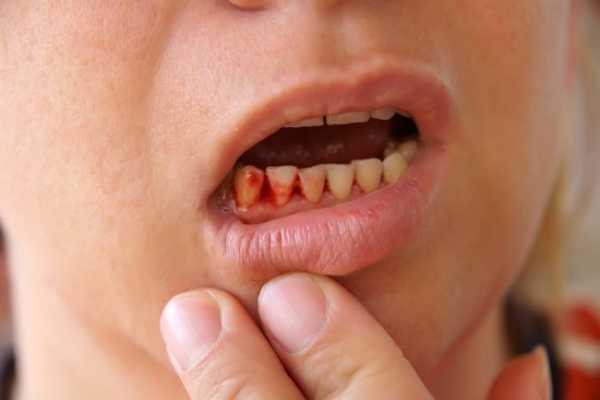
Кровотечения при Синдроме Вискотта-Олдрича отличаются длительностью и рецидивирующим характером
Впервые заболевание было описано в начале прошлого века. Доктор Вискотт описал троих братьев, рождённых от одной матери, со специфическими симптомами — кровавой диареей, поражениями кожи и ушными инфекциями. Через несколько десятков лет доктор Олдрич доказал, что этот синдром наследуется по материнской линии вместе с аномальным геном.
Причины возникновения
Заболевание относится к крайне редким патологиям и встречается в 4–10 случаях на миллион. Синдром Вискотта-Олдрича поражает исключительно мужчин — женщины являются носителями дефектного гена, но клинические проявления болезни у них отсутствуют. Место проживания или какие-либо другие факторы не оказывают влияния на заболеваемость, так как патология имеет генетическое происхождение. Риск заболеть при наличии соответствующего семейного анамнеза составляет 50% – мать-носительница может передать аномальный ген половине своих детей мужского пола, а половина девочек может стать его носительницами.

Для беременных иногда проводят забор ворсин хориона (оболочки плода) на 9–12 неделе вынашивания плода
В основе механизма развития болезни лежит мутация гена WASp, расположенного на так называемом коротком плече Х-хромосомы. В результате в организме человека перестаёт вырабатываться белок WASp — вещество, которое отвечает за взаимодействие клеток иммунной системы, вследствие чего ее работа нарушается.
Симптомы
Симптомы синдрома Вискотта-Олдрича начинают проявляться сразу после рождения или в возрасте 4–8 месяцев, когда материнские иммуноглобулины перестают защищать организм ребёнка. Симптомокомплекс заболевания включает три основных группы признаков — кожные поражения, тромбоцитопения (нарушения, вызванные низким уровнем тромбоцитов) и проявления иммунодефицита.

Диагностикой и лечением заболевания занимаются аллерголога-иммунологи, гематологи, медицинские генетики
Характерные проявления патологии:
- Частые инфекционные болезни, которые развиваются вследствие нарушения работы иммунной системы. Они связаны с недостаточной выработкой белковых веществ, принимающих участие в уничтожении чужеродных агентов.
- Заболевания, вызванные патогенными микроорганизмами, которые рецидивируют и тяжело поддаются лечению. Обычно в качестве возбудителей выступают бактерии и вирусы, которые входят в состав условно-патогенной микрофлоры организма, то есть постоянно присутствуют в нём, но не проявляют себя при нормальной работе иммунитета. У младенцев чаще всего наблюдаются отиты и ушные инфекции, вызванные стафилококками, стрептококками, синегнойной палочкой, вирусом герпеса, грибками и так далее. В старшем возрасте у больных развиваются поражения слизистых оболочек ЖКТ, печени, мочеполовой и других систем, вызванные теми же патогенными микроорганизмами.
- Кровотечения возникают из-за нарушения гемостаза и снижения количества тромбоцитов. Они могут быть как наружными (из слизистых оболочек полости рта и носоглотки, небольших порезов и травм), так и внутренними — образование гематом, петехий и синяков, кровавый понос.
- Экзема представляет собой различные дефекты, которые появляются на коже больного — сыпь, пятна, шелушащиеся участки и эрозии, сопровождающиеся зудом и дискомфортом.
Основная особенность симптомов болезни заключается в том, что они тяжело поддаются лечению и постоянно рецидивируют, иногда вне зависимости от внешних факторов — например, синяки и гематомы на коже могут появляться при отсутствии механических травм.
Выраженность проявлений может быть разной — у больных с полным отсутствием выработки белка WASp патология протекает тяжело, с ярко выраженными признаками, а в случаях когда его продукция снижена, клиническое течение чаще всего лёгкое.
Диагностика

На сегодняшний день разрабатываются методы генной терапии (изменение генотипа пораженных стволовых клеток путем внесения нужных генов различными посредниками (при помощи вирусов))
Диагноз при синдроме Вискотта-Олдрича ставится на основе комплексного исследования, которое включает:
- сбор симптоматики и анамнеза, в том числе семейного — наличие у больного характерных симптомов, их интенсивность и продолжительность, состояние здоровья родителей;
- внешний осмотр — выявляет наружные признаки нарушения свёртываемости крови и поражения кожных покровов — синяки, точечные кровоизлияния, сыпь, экзему;
- анализ крови — позволяет определить снижение количества тромбоцитов и изменение их размера, снижение концентрации иммуноглобулинов — белковых соединений, выступающих показателями нормальной работы иммунной системы;
- генетическое исследование — для выявления патологических изменений в гене WASp.
Если женщина знает о том, что является носительницей дефектного гена, диагностику нужно провести ещё в период вынашивания ребёнка. Для этого следует обратиться к специалисту-генетику и пройти специальные обследования — например, забор ворсин хориона для выявления генетических дефектов в клетках плода. В случаях когда у новорождённого присутствуют признаки синдрома Вискотта-Олдрича, нужно обратиться к аллергологу-иммунологу и гематологу.
Лечение
Лечение синдрома Вискотта-Олдрича может быть симптоматическим, то есть направленным на облегчение состояния больного и улучшение качества его жизни, а также радикальным — чаще всего используется пересадка костного мозга.

Вакцинация детей с синдром Вискотта-Олдрича является обязательной
Варианты терапии:
- Консервативная. Включает в себя приём стероидных препаратов и антигистаминных средств для устранения симптомов экземы, применение антибиотиков, антимикробных и иммуномодулирующих препаратов для лечения инфекционных болезней.
- Трансфузионная. Переливание компонентов крови (тромбоцитов, эритроцитарной массы, плазмы, факторов роста) помогает предотвратить кровотечение и развитие инфекций.
- Трансплантация стволовых клеток. Пересадка костного мозга — ключевой и наиболее эффективный способ лечения синдрома Вискотта-Олдрича, который позволяет добиться стойкой, длительной ремиссии, улучшить качество и продолжительность жизни больного.
Терапия заболевания должна быть длительной — даже в случае успешного оперативного вмешательства пациенту придётся принимать поддерживающие препараты. Кроме лечения, больным с этим диагнозом необходимо вести особый образ жизни — избегать травм, чрезмерной физической активности, инфекционных и простудных заболеваний, тщательно соблюдать личную гигиену.
Прогноз и осложнения
Синдром Вискотта-Олдрича, особенно в тяжёлой форме, способен повлечь за собой большое количество осложнений и неприятных последствий, в число которых входят:
- тяжёлые пневмонии;
- септические поражения организма;
- аутоиммунные нарушения;
- онкологические болезни, чаще всего лимфомы и лейкозы.
Прогноз жизни для больных зависит от особенностей клинического течения заболевания и других факторов. Больные с лёгкой формой, чётко соблюдающие врачебные рекомендации, могут прожить долгую жизнь, хотя и с некоторыми ограничениями. Вместе с тем у 10% взрослых больных развиваются злокачественные заболевания кроветворной системы, которые ведут к тяжёлым последствиям и летальному исходу.
Синдром Вискотта-Олдрича относится к заболеваниям, которые нельзя предотвратить или полностью вылечить, но избежать тяжёлых последствий и улучшить качество жизни больного вполне возможно. Для этого нужно внимательно относиться к собственному здоровью, планированию беременности и внимательно следить за состоянием ребёнка, а при развитии тревожных симптомов обращаться к врачу.
Оцените статью: Поделитесь с друзьями!Синдром вискотта олдрича у детей что это такое
Синдром Вискотта-Олдрича: причины развития, признаки и проявления, диагноз, как лечить
Содержание:
Синдром Вискотта-Олдрича (СВО) — наследственная патология, обусловленная дефицитом особого белка WASp, обеспечивающего взаимодействие между клетками крови. Он принимает непосредственное участие в процессе свертывания крови при повреждении кровеносных сосудов, а также в поддержании иммунной защиты организма от патогенных и условно-патогенных микробов. СВО относится к группе первичных иммунодефицитных состояний, обусловленных поражением Т- и В — лимфоцитов, продуцирующих антитела. Дефицит тромбоцитов приводит к повышенной кровоточивости и массивной кровопотере. Заболевание проявляется триадой симптомов — экземой, первичным иммунодефицитом и тромбоцитопенией.
СВО является редкой Х-сцепленной рецессивной патологией, при которой женщины считаются носителями мутантного гена. При этом сами они не болеют, а передают поврежденный ген детям. Это легко объяснить наличием у них здоровой аналогичной хромосомы, которая не позволяет недугу развиться. У сыновей болезнь проявится клинически, а дочери становятся носителями мутантного гена.
Принципы наследования СВО
Синдром впервые был описан в 1937 году немецким педиатром Вискоттом. Он наблюдал за тремя братьями, у которых имелись проявления тромбоцитопении, рецидивирующих инфекций уха, экземы. В 1954 году детский врач из Америки Олдрич определили характер наследования заболевания. Спустя много лет был выявлен ген, мутации которого приводят к синдрому. В 50-х и 60-х годах 20-го века синдром вошел в список первичных иммунодефицитов на основании признаков иммунной недостаточности у больных.
Патология иммунной системы развивается в период эмбриогенеза. Врожденный иммунодефицит приводит к развитию тяжелых инфекционных недугов. Он часто сочетается с тромбоцитопенией и экземой на лице, конечностях или всего тела. У больных СВО значительно повышается риск развития злокачественных опухолей и аутоиммунных нарушений. Дети с данной патологией в наибольшей степени подвержены бактериальной, грибковой или вирусной инфекции.
СВО встречается у 4-10 из 1 миллиона родившихся живыми детей. Географический фактор при этом не имеет значения. Страдают патологией исключительно мужчины. Женщины же являются гетерозиготными носителями патологии.
ребенок с синдромом Вискотта-Олдрича
Выделяют три формы СВО:
- Легкая форма – микротромбоцитопения, иммунодефицит, отсутствие экземы, нечастые инфекции, проходящие без осложнений;
- Среднетяжелая и тяжелая формы фактически мало чем отличаются друг от друга и проявляются экземой, плохо поддающимися лечению аутоиммунными, инфекционными и онкологическими заболеваниями.
Этиология
СВО — генетически детермированная патология, в основе которой лежит мутация гена, ответственного за синтез белка, функция которого до конца не известна. Его дефект, недостаток или полное отсутствие приводят к нарушению иммунной защиты и работы свертывающей системы крови. У больных с СВО в результате мутации развивается иммунодефицит и тромбоцитопения.
Иммунодефицит обусловлен неспособностью иммунокомпетентных клеток воспринимать информацию об антигенах и продуцировать антитела. Подобные расстройства связаны с нарушениями белкового обмена. При снижении иммунной защиты организма возникают острые инфекции различной этиологии у людей с СВО.
Поражение белковых молекул, участвующих в процессе тромбоцитообразования, приводит к снижению количества и ухудшению качества этих клеток. В результате возникают частые кровотечения, экземы, язвы на коже. Аномально уменьшенные в размерах тромбоциты перестают выполнять свои функции в полном объеме и разрушаются в селезенке. Это приводит к тромбоцитопении и спленомегалии. У больных часто появляются синяки и петехии, кровотечения из носа и десен, кровавый понос. Причиной плохого самочувствия у таких больных становится анемия.
Основные типы мутации генов при СВО:
- Мутации гена, приводящие к образованию усеченного белка, вызывают СВО с тяжелым течением и ярко выраженной симптоматикой. Когда выработка белка полностью прекращается, возникает «классическая», самая опасная, форма болезни.
- Мутации гена, кодирующего нормальную длину белка, приводят к развитию типичной формы СВО. Выработка некоторого количества измененного белка проявляется среднетяжелым течением синдрома.
Оба типа генетических мутаций могут привести к аутоиммунным заболеваниям и злокачественным новообразованиям у больных.
Мутантный ген, носителем которого является беременная женщина, передается по наследству детям следующими способами:
- в 25% случаев беременности рождается здоровая дочь-носительница,
- в 25% – здоровая дочь,
- в 25% – больной сын,
- в 25% – здоровый сын.
СВО проявляется симптомами атопического дерматита и геморрагического синдрома. Дефицит В- и Т-лимфоцитов приводит к часто повторяющимся и тяжело протекающим инфекционным процессам. У больных развиваются заболевания ЛОР-органов, бронхов и легких, кожи, почек и пищеварительного тракта.
Чаще всего организм поражают инфекции, вызванные условно-патогенными микроорганизмами — золотистым стафилококком, гемолитическим стрептококком, синегнойной палочкой, некоторыми энтеробактериями, а также патогенными грибками и вирусами. Бороться с оппортунистическими инфекциями очень сложно, поскольку аутофлора, вызывающая воспаление, присутствует на всей поверхности тела человека. У больных часто рецидивирует герпетическая инфекция, проявляющаяся ангинами, стоматитом, поражением половых органов. Активируются вирусы Эпштейна-Барр, цитомегаловирус. Организм не может противостоять бактериальной атаке, поскольку имеется неполноценность белков, подавляющих размножение микробов и уничтожающих их.
Симптомы СВО впервые проявляются на первых месяцах жизни. С возрастом они прогрессируют. У мальчиков с синдромом Вискотта – Олдрича возникают следующие клинические признаки:
- наружные кровотечения при травматическом повреждении, кровоточивость десен, носовые кровотечения;
- внутренние кровотечения — желудочно-кишечное, подкожное, внутрисуставное;
- признаки анемии — головокружение, слабость, упадок сил, тошнота;
- гематурия, кровь в кале, кровавая рвота;
- боль в суставах;
- экзема — поражение кожи аллергической природы с образованием гиперемированных пятен, язв и эрозий на лице, конечностях и ягодицах;
- зудящие шелушащиеся высыпания на коже, напоминающие клиническую картину атопического дерматита и возникающие в межсезонье в ответ на аллергенный агент.
Проявления синдрома Вискотта-Олдрича
На фоне кровопотери снижается общий гемоглобин и возникает гипоксия тканей. Компенсаторно повышается давление и увеличивается нагрузка на миокард и стенки сосудов. Так развивается сердечная недостаточность. Эндотелий сосудов легко повреждается и плохо восстанавливается, приводя к опасным для жизни сосудистым катастрофам.
Тяжесть клинических проявлений может варьироваться от проходящей тромбоцитопении с незначительными геморрагическими признаками до тяжелого заболевания с выраженным симптомами инфекционных и аутоиммунных нарушений. Симптомы СВО ухудшают общее состояние больных и выбивают их из привычного ритма жизни.
Дети, дожившие до 10 лет, обычно страдают хотя бы одним, а чаще несколькими, аутоиммунными заболеваниями – васкулитом, гемолитической анемией, полиартритом. Постоянные васкулиты приводят к гибели пациентов от острой коронарной или мозговой недостаточности. Подавление активности иммунных клеток или снижение их количества приводит к развитию онкозаболеваний — лейкоза или лимфомы. Осложнениями частых инфекционных заболеваний становятся тяжелые пневмонии и сепсис.
Диагностические мероприятия
Синдром Вискотта-Олдрича подозревают у всех мальчиков с кровотечениями и врожденной тромбоцитопенией. Признаки острых инфекций и аутоиммунных расстройств могут отсутствовать или быть слабо выраженными.
16-летний мальчик с СВО
- Чтобы подтвердить или опровергнуть предполагаемый диагноз, специалисты собирают анализ и в
Вискотта-Олдрича синдром | Фонд «Подари жизнь»
Суть болезни
Синдром Вискотта-Олдрича (СВО) – редкое наследственное заболевание, которое характеризуется сочетанием трех симптомов: кожной экземы, тромбоцитопении (то есть низкого уровня тромбоцитов) и иммунодефицита. Таким образом, СВО относится к первичным иммунодефицитам. Кроме того, у больных СВО значительно повышен риск развития злокачественных опухолей и аутоиммунных нарушений.
Частота встречаемости и факторы риска
СВО – редкое заболевание; частота его оценивается как 4-10 случаев на 1 миллион живых новорожденных. Встречается во всех регионах.
Так как СВО характеризуется Х-сцепленным наследованием, болеют почти исключительно мальчики (описаны лишь единичные случаи, где атипичная форма этого синдрома предполагается у девочек). Заболевание может быть унаследовано с вероятностью 50% в случае, если мать мальчика является носительницей дефектного гена; при этом сама она клинически здорова. Семьям, где уже были случаи СВО у детей, рекомендована консультация генетика.
Признаки и симптомы
Симптомы СВО обычно начинают проявляться уже в течение первых месяцев жизни. С возрастом они, как правило, прогрессируют.
Тромбоциты при СВО имеют уменьшенный размер, а их функция нарушена. Эти аномальные тромбоциты разрушаются в селезенке; в результате уровень тромбоцитов снижается, а размер селезенки может увеличиваться. Тромбоцитопения проявляется возникновением синяков, подкожных кровоизлияний (петехий) и кровотечений – например, нередки кровавый понос, кровотечения из носа и десен. Из-за этого у больного может также развиться анемия.
В большинстве случаев одним из симптомов является зудящая кожная сыпь, хотя у некоторых больных она может быть слабо выраженной или даже отсутствовать.
Иммунодефицит связан как с нарушением функции В-лимфоцитов, так и с дефицитом Т-лимфоцитов, то есть речь идет о комбинированном иммунодефиците. Его следствием являются упорные, часто повторяющиеся бактериальные, вирусные и грибковые инфекции, которые могут поражать верхние и нижние дыхательные пути, пазухи носа, уши, глаза, кожу, слизистые оболочки, желудочно-кишечный тракт, мочевыводящие пути.
У больных СВО существенно повышен риск возникновения злокачественных заболеваний, прежде всего лимфом и лейкозов; с возрастом этот риск увеличивается. В ряде случаев развиваются и аутоиммунные нарушения, такие как васкулит (поражение кровеносных сосудов) или гемолитическая анемия.
Конкретные генетические дефекты (мутации), вызывающие СВО, могут различаться у разных детей. Соответственно, тяжесть протекания болезни зависит от того, каким именно дефектом она вызвана в каждом случае. Все эти дефекты касаются гена особого белка WASP (Wiskott-Aldrich syndrome protein), который выполняет определенные функции в клетках крови. Если в результате мутации выработка этого белка почти полностью прекращается, то наблюдаются «классические», более тяжелые формы болезни; если же возможна выработка некоторых количеств измененного белка, то болезнь протекает не так тяжело.
Диагностика
СВО диагностируется на основе клинических симптомов, результатов клинического анализа крови, микроскопического
Белок синдрома Вискотта — Олдрича — Википедия
Материал из Википедии — свободной энциклопедии
Белок синдрома Вискотта — Олдрича (англ. The Wiskott–Aldrich Syndrome Protein, WASp) состоит из 502 остатков аминокислот[1] и имеет сложное доменное строение. Он экспрессируется в гемопоэтических клетках, играет важную роль в реорганизации цитоскелета, сигнальной трансдукции и апоптозе[2].
Ген WAS, мутации в котором приводят к развитию синдрома Вискотта — Олдрича, локализован на коротком плече Х-хромосомы в области Хp11.4 — Хp11.21. Точнее, ген WAS расположен от до 48,691,426-й пары оснований на Х-хромосоме[3]. Картирование гена было произведено в 1994 году, было установлено, что ген WAS состоит из 12 экзонов и 11 интронов[4] общей протяженностью приблизительно 1800 пар нуклеотидов[5][6].
На С-конце белка WASp, в непосредственной близости один от другого, расположены домены связывания мономерного актина и Агр2/3-комплекса. Это обеспечивает пространственное сближение этих молекул и облегчает процесс полимеризации актина. На N-конце и в центральной части белка находятся различные регуляторные домены (Wh2, GBD, Роlу-Рrо-участок), обеспечивающие взаимосвязь со множеством специальных белковых молекул и фосфолипидов мембран. Таким образом, белок WASp может служить посредником при взаимодействии различных специфических факторов с мембранными структурами и участвовать в процессе полимеризации актина[7][8].
Возникновение мутации в гене, ответственном за синтез белка WASp, приводит к появлению дефектной формы белка или к полному его отсутствию, что влечёт за собой развитие нарушений иммунитета и гемостаза. Известно не менее 350 мутаций[3] в гене WAS, ответственных за развитие Х-сцепленного синдрома Вискотта — Олдрича. Основное их количество приходится на нуклеотидные замены (миссенс-/нонсенс-мутации), мутации сплайсинга и малые делеции. Выявляются так же малые инсерции и большие делеции[9].
У больных с миссенс-мутациями, как правило, наблюдается лёгкое течение заболевания, проявляющееся преимущественно тромбоцитопенией. Для пациентов с различными делециями, вставками нуклеотидов, нонсенс-мутациями и мутациями сайтов сплайсинга характерны более тяжёлые клинические проявления[10][11].
- ↑ P42768[1-502], Wiskott-Aldrich syndrome protein, Homo sapiens
- ↑ Синдром Вискотта-Олдрича. Резюме (рус.) (pdf). orpha.net (2013). Дата обращения 7 января 2017.
- ↑ 1 2 A service of the U.S. National Library of Medicine: Genetics Home Reference
- ↑ WAS — Wiskott-Aldrich syndrome (eczema-thrombocytopenia) Архивировано 1 февраля 2014 года., Resource of Asian Primary Immunodeficiency Diseases (RAPID)
- ↑ Derry J. M., Ochs H. D., Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. (англ.) // Cell. — 1994. — Vol. 78, no. 4. — P. 635—644. — PMID 8069912.
- ↑ chrX:48542185-48549817 7,633 bp. UCSC Genome Browser on Human Feb. 2009 (GRCh47/hg19) Assembly
- ↑ Гусев Н. Б. Движение немышечных клеток и реорганизация актиновых микрофиламентов // Соросовский образовательный журнал. — 2001. — Т. 7, № 7. — С. 9—16.
- ↑ Thrasher A. J., Burns S. Wiskott-Aldrich syndrome: a disorder of haematopoietic cytoskeletal regulation. (англ.) // Microscopy research and technique. — 1999. — Vol. 47, no. 2. — P. 107—113. — doi:10.1002/(SICI)1097-0029(19991015)47:2<107::AID-JEMT3>3.0.CO;2-H. — PMID 10523789.
- ↑ База данных: HGMD — Human Gene Mutation Database.
- ↑ Lemahieu V., Gastier J. M., Francke U. Novel mutations in the Wiskott-Aldrich syndrome protein gene and their effects on transcriptional, translational, and clinical phenotypes. (англ.) // Human mutation. — 1999. — Vol. 14, no. 1. — P. 54—66. — doi:10.1002/(SICI)1098-1004(1999)14:1<54::AID-HUMU7>3.0.CO;2-E. — PMID 10447259.
- ↑ Lutskiy M. I., Rosen F. S., Remold-O'Donnell E. Genotype-proteotype linkage in the Wiskott-Aldrich syndrome. (англ.) // Journal of immunology (Baltimore, Md. : 1950). — 2005. — Vol. 175, no. 2. — P. 1329—1336. — PMID 16002738.
Сермягина И. Г., Забненкова В. В., Кондратенко И. В., Поляков А. В., ДНК-диагностика синдрома Вискотта-Олдрича // Медицинская генетика, 2009 г., т. 8, N6(84), с. 34—39 (недоступная ссылка)
Симптомы синдрома Вискотта-Олдрича | Компетентно о здоровье на iLive
Тяжесть симптомов заболевания у больных с синдромом Вискотта-Олдрича варьирует от интермиттирующей тромбоцитопении с минимальными геморрагическими проявлениями до тяжелого заболевания с выраженным инфекционным и аутоиммунным синдромами. Таким образом, на настоящий момент не удалось установить четкой корреляции между тяжестью заболевания и типом мутации. Расхождения между несколькими группами исследователей могут объясняться отсутствием четкой классификации WAS и, как следствие этого, исследователи классифицируют больных со сходной выраженностью болезни по разному. Tew не менее, в целом большинство миссенс мутаций во 2 экзоне сопровождаются легким течением заболеваний, нонсенс и СРС мутации ведут к тяжелому синдрому Вискотта-Олдрича.
Геморрагический синдром
Средний возраст постановки диагноза синдрома Вискотта-Олдрича по данным исследования 1994 года составляет 21 месяц, и у 90% больных геморрагический синдром присутствует в момент постановки диагноза. Поскольку тромбоцитопения, как правило, отмечается с рождения, заболевание может манифестировать кровотечениями из пупочной ранки, а также такими симптомами, как мелена, эпистаксис, гематурия, петехиальная сыпь, а также жизнеугрожающими внутричерепными и гастроинтестинальными кровотечениями. В 1994 г, кровотечения были отмечены как основная причина летальности при синдроме Вискотта-Олдрича.
Больным с синдромом Вискотта-Олдрича нередко ставится диагноз идиопатической тромбоцитопенической пурпуры (ИТП), что значительно оттягивает постановку настоящего диагноза.
У некоторых больных с синдромом Вискотта-Олдрича тромбоцитопения и геморрагические проявления являются единственными симптомами заболевания, и на протяжении многих лет, до выявления гена, ответственного за данный синдром, этих больных относили в группу Х-сцепленной тромбоцитопении. При более тщательном обследовании у некоторых из них удавалось выявить лабораторные нарушения иммунного ответа при отсутствии или минимальных клинических проявлениях иммунодефицита.
Экзема или атопический дерматит различной степени выраженности проявляется, как правило, на первом году жизни и часто сопровождается местным инфицированием, У больных с легким течением WAS экзема может отсутствовать или носить легкий, преходящий характер.
Инфекционные проявления
У большинства больных синдромом Вискотта-Олдрича с возрастом проявляются прогрессирующие признаки иммунодефицита. Вследствие нарушений гуморального и клеточного иммунитета, у больных со среднетяжелым или тяжелым течением синдрома Вискотта-Олдрича наблюдаются частые инфекции, которые нередко встречаются в первые шесть месяцев жизни. Из них наиболее распространенны воспаление среднего уха (78%), синуситы (24%) и пневмония (45%). То же ретроспективное исследование показало, что у 24% больных наблюдался сепсис, у 7% менингит и инфекции ЖКТ у 13%. Наиболее частыми возбудителями являются Н. influenzae, S. pneumoniae, P. carinii, C. albicans. Реже встречаются вирусные инфекции, которые включают ветрянку и герпетические инфекции. Грибковые заболевания встречаются редко. У больных с легким течением синдрома Вискотта-Олдрича упоминания о частых инфекциях могут отсутствовать.
Аутоиммунные заболевания
По данным Sullivan аутоиммунные нарушения наблюдаются у 40% больных синдромом Вискотта-Олдрича. Наиболее часто встречаются гемолитическая анемия, васкулиты и поражение почек. Аутоиммунные нарушения характерны для тяжелого течения болезни. Некоторые больные развивают более одного аутоиммунного заболевания. Часто больные WAS развивают иммунную тромбоцитопению, сопровождающуюся повышенным уровнем тромбоцитарного IgG. У больных синдромом Вискотта-Олдрича, имеющих нормальный уровень тромбоцитов как следствие спленэктомии, подчас наблюдается повторное снижение числа тромбоцитов в результате вторичного аутоиммунного процесса.
Злокачественные новообразования
Злокачественные новообразования чаще развиваются у взрослых или подростков с синдромом Вискотта-Олдрича, но могут встречаться и у детей. Средний возраст развития злокачественных новообразований у больных синдромом Вискотта-Олдрича составляет 9,5 лет. Ранее у больных WAS старше 5 лет частота опухолевых заболеваний составляла в среднем 18-20%. С увеличением продолжительности жизни больных синдромом Вискотта-Олдрича вследствие улучшения медицинской помощи возросла пропорция больных, у которых развиваются опухолевые заболевания. Большинство опухолей имеют лимфоретикулярное происхождения, среди них чаще всего встречаются неходжкинские лимфомы, вто время, как типичные для детского возраста нейробластома, рабдомиосаркома, саркома Юинга и др. отсутствуют. Лимфомы часто имеют экстранодальную локализацию и характеризуются неблагоприятным прогнозом.
Лабораторная патология
Как было сказано выше, наиболее постоянным проявлением синдрома Вискотта-Олдрича является тромбоцитопения со снижением объема тромбоцитов. Сниженный объем тромбоцитов является практически уникальным симптомом, позволяющим проводить дифференциальный диагноз с другими тромбоцитопениями. Проводить определение функциональных характеристик тромбоцитов в условиях клинической лаборатории не рекомендуется, так как это исследование усложнено уменьшенным объемом тромбоцитов больных WAS.
Иммунные нарушения при синдроме Вискотта-Олдрича включают е себя нарушения как гуморального, так и клеточного звена. Нарушения Т-клеточного иммунитета включают в себя в первую очередь, лимфопению, которая отмечается у больных WAS с раннего возраста. В большей степени у больных снижены CD8 лимфоциты. Кроме того, у больных WAS отмечаются снижение реакции на митогены, снижение пролиферации в ответ на стимуляцию аллогенными клетками и моноклинальными антителами к CD3, нарушение реакции гиперчувствительности замедленного типа в ответ на специфические антигены. Реакции гиперчувствительности замедленного типа нарушены у 90% больных. В гуморальном звене отмечается умеренное снижение В-пимфоцитов, снижение уровня IgM, нормальный или сниженный уровень IgG, повышение IgA и ГдЕ. Интересной особенностью иммунного статуса больных WAS является относительное и абсолютное повышение натуральных киллеров. Существуют данные, что этот факт имеет патогенетическое значение.
Для синдрома Вискотта-Олдрича также характерна неспособность больных синтезировать антитела к пол исахаридным антигенам. Впервые этот дефект был описан как отсутствие у этих больных изогенагглютининов. Позже было показано, что больные с синдромом Вискотта-Олдрича неспособны продуцировать антитела в ответ на такие антигены, как пневмококковые полисахариды, липополисахарид VI E.coli антигены сальмонеллы.
Стандартные исследования нейтрофильного и макрофагального звеньев иммунитета, включавшие исследования подвижности нейтрофилов, фагоцитарного ответа, высвобождения гранул не выявили нарушений. Существуют сообщения о нарушении хемотаксиса нейтрофилов и моноцитов.
Синдром Вискотта-Олдрича: описание заболевания
Синдром Вискотта-Олдрича относится к наследственным недугам, которым болеют только мальчики. Проявляется он уже в раннем детстве и, как правило, приводит к смерти через пять-десять лет.
Впервые данное заболевание было описано в 1937 году немецким педиатром Вискоттом. Он наблюдал его у трех родных братьев, которые страдали непрекращающимся кровавым поносом, экземой и постоянным воспалением уха, хотя четыре сестры были абсолютно здоровы. Уже в 1954 году американский педиатр по фамилии Олдрич установил, что данная болезнь наследуется как Х-сцепленный рецессивный признак.
Недуг передается мальчикам, а носителем мутирующей хромосомы являются женщины. Географический фактор на распространенность проблемы не влияет.
Симптомы и течение заболевания
Синдром Вискотта-Олдрича связан с пониженным количеством тромбоцитов. В первые годы жизни у больного проявляются следующие симптомы: плохая свертываемость крови, непрекращающиеся экземы, кровавый понос. Впоследствии наблюдается первичное иммунодефицитное состояние. Из-за недостатка Т- и В-лимфоцитов человек подвержен всевозможным вирусным и бактериальным заболеваниям. Иммунодефицитные состояния у детей провоцируют постоянное воспаление среднего уха, пневмонию, синусит, водянку и многие другие тяжелые заболевания. У страдающих данным заболеванием риск развития онкологии намного выше. Следует отметить, что злокачественным новообразованиям чаще подвержены взрослые.
У страдающих синдромом Вискота-Олдрича очень часто наблюдают только геморрагический синдром, что приводит к неправильной постановке диагноза. В таких случаях только после анализа ДНК, при котором можно выявить ген, ответственный за данный синдром, назначают правильное лечение.
Диагностика
- Полный анализ крови на выявление снижения количества тромбоцитов.
- Исследование мазка крови на структурную неполноценность тромбоцитов.
- Генетический анализ (проводится для обнаружения мутаций в гене, отвечающем за строение белка в крови).
- Определение уровня иммуноглобулина.
- Проведение пренатальной диагностики, чтобы выявить синдром Вискотта-Олдрича на ранних стадиях беременности.
- Определение в крови уровня белка Вискотта-Олдрича.
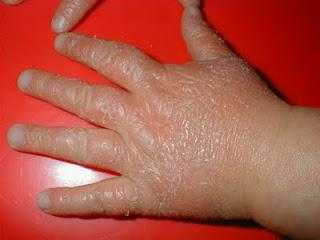
Лечение
К сожалению, современная наука пока не нашла лекарств от этого тяжелого заболевания. Известно, что тромбоциты разрушаются в селезенке, поэтому у больных, которым был поставлен диагноз "синдром Вискотта-Олдрича", удаляли данный орган. И пациенты начинали чувствовать себя намного лучше. Постоянное переливание иммуноглобулина и назначение соответствующих антибиотиков также улучшает общее состояние больного. В настоящее время практикуется внедрение больным в костный мозг здоровых стволовых клеток. Но пока этот метод проводится только экспериментально. Также парам, у которых в семье наблюдался данный синдром, рекомендуется перед планированием беременности сдать все необходимые анализы.
Болезнь для мальчиков – Газета Коммерсантъ № 148 (4203) от 14.08.2009
Яну год. У него синдром Вискотта-Олдрича — врожденный иммунодефицит, который, подобно гемофилии, передается по женской линии, а болеют мальчики. Дети с этим синдромом умирают в возрасте, как правило, от двух до пяти лет. Умирают от простейших инфекций, или от абсцесса легкого, или от внутренних кровотечений. Единственное лечение — пересадка чужого костного мозга. Шансы пятьдесят на пятьдесят. Чем раньше, тем лучше. Тем меньше инфекций принесет внутри себя ребенок в стерильный бокс, где делают трансплантацию.
У маленького Яна был старший брат Егор. Он умер двухлетним...
Нет, не с этого нужно начинать.
У бабушки Яна Корченкова Светланы Дмитриевны, кроме дочери Кати, был еще сын, который умер во младенчестве. Непонятно было, от чего умер. Светлана Дмитриевна была женою военного. Муж служил где-то на Дальнем Востоке. И там, в расположении военной части, военврач только развел руками, когда сын одного из офицеров ни с того ни с сего истек кровью и умер. До ближайшей больницы ехать было так долго, что кровью истекают быстрее. Синдром Вискотта-Олдрича был уже открыт и объяснен, но военврачу неизвестен хотя бы потому, что военврачи специализируются на сортировке раненых, а не на гематологии и педиатрии. Трансплантаций костного мозга в России тогда не делали. Ребенка все равно нельзя было бы спасти, даже если бы военврач про синдром Вискотта-Олдрича знал. Доктор просто констатировал смерть и причиною смерти записал внутреннее кровотечение.
Семь лет назад у Светланы Дмитриевны родился внук Егор. Это было уже в Петербурге. Российская детская гематология к тому времени развилась уже настолько, что синдром Вискотта-Олдрича ребенку диагностировали уверенно. И сказали, что ребенок умрет года в два. Так оно и случилось.
Первый год жизни Егора был относительно безоблачным. Ребенок как ребенок. Второй год прошел в каретах скорой помощи, в реанимационных отделениях, с катетерами в венах, с зондами во рту.
Мальчику ничем не могли помочь. Он болел всеми болезнями сразу. Каждый месяц, даже если не находилось какой-нибудь конкретной инфекции, которая привела бы малыша в отделение интенсивной терапии, то тогда просто кровь шла носом или горлом и никак не останавливалась.
В последние пять или десять госпитализаций Егора врачи уже прямо говорили матери и бабушке, что ребенок умрет, и умрет скоро. В последний раз его выписали из больницы не потому, что ему стало лучше, а потому, что у него был день рождения. Ему исполнялось два года. Последний праздник, который можно было устроить дома с подарками, игрушками и тортом.
Егор не чувствовал себя хорошо ни в самый день рождения, ни после него, но кое-как все еще жил. Целый месяц.
Наконец однажды у него пошла кровь горлом, и Светлана Дмитриевна вызвала скорую, хотя дочь и говорила, что скорую не нужно, скорая не поможет, хватит мучить ребенка, надо дать ему умереть.
Скорая приехала. Врачи настаивали на немедленной госпитализации, но мать сказала: "Вы спасете его? Вы будете его только мучить. Оставьте его в покое". Они сидели и смотрели, как у ребенка маленькими, замедляющимися волнами вытекает кровь изо рта, как ребенок слабеет, затихает и умирает.
Первая (неудачная) трансплантация костного мозга ребенку с синдромом Вискотта-Олдрича была проведена в России через год после смерти Егора. В дальнейшем в половине случаев трансплантации излечивали таких детей.
После смерти Егора его мать начала всерьез пить. Второго сына Яна зачала и родила, не понимая, что делает. Совершенно не заботилась о ребенке и была лишена родительских прав. Опеку над мальчиком оформила бабушка Светлана Дмитриевна. Это третий у нее на руках ребенок с синдромом Вискотта-Олдрича.
Это первый в роду ребенок с с