Биоинформатика что это такое
Биоинформатика — Википедия
 Карта Х хромосомы человека (с сайта NCBI). Сборка человеческого генома — это одно из величайших достижений биоинформатики.
Карта Х хромосомы человека (с сайта NCBI). Сборка человеческого генома — это одно из величайших достижений биоинформатики. Биоинформа́тика — совокупность методов и подходов[1], включающих в себя:
- математические методы компьютерного анализа в сравнительной геномике (геномная биоинформатика).
- разработку алгоритмов и программ для предсказания пространственной структуры биополимеров (структурная биоинформатика).
- исследование стратегий, соответствующих вычислительных методологий, а также общее управление информационной сложности биологических систем[2].
В биоинформатике используются методы прикладной математики, статистики и информатики. Биоинформатика используется в биохимии, биофизике, экологии и в других областях.
Наиболее часто используемыми инструментами и технологиями в этой области являются языки программирования Java, C#, Perl, C, C++, Python, R; язык разметки — XML; базы данных — SQL; программно-аппаратная архитектура параллельных вычислений — CUDA; пакет прикладных программ для решения задач технических вычислений и одноимённый язык программирования, используемый в этом пакете — MATLAB, и электронные таблицы.
Биоинформатика стала важной частью многих областей биологии. В экспериментальной молекулярной биологии методы биоинформатики, такие как создание изображений и обработка сигналов, позволяют получать полезные результаты из большого количества исходных данных. В области генетики и геномики, биоинформатика помогает в упорядочивании и аннотировании геномов и наблюдаемых мутаций. Она играет роль в анализе данных из биологической литературы и развитии биологических и генетических онтологий по организации и запросу биологических данных. Она играет роль в анализе гена, экспрессии белка и регуляции. Инструменты биоинформатики помогают в сравнении генетических и геномных данных и, в целом, в понимании эволюционных аспектов молекулярной биологии. В общем виде, она помогает анализировать и каталогизировать биологические пути и сети, которые являются важной частью системной биологии. В структурной биологии, она помогает в симуляции и моделировании ДНК, РНК и белковых структур, а также молекулярных взаимодействий.
История[править | править код]
Опираясь на признание важной роли передачи, хранения и обработки информации в биологических системах, в 1970 году Полина Хогевег ввела термин «биоинформатика», определив его как изучение информационных процессов в биотических системах[3][4]. Это определение проводит параллель биоинформатики с биофизикой (учение о физических процессах в биологических системах) или с биохимией (учение о химических процессах в биологических системах)[3].
В начале «геномной революции» термин «биоинформатика» был переоткрыт и обозначал создание и техническое обслуживание базы данных для хранения биологической информации
Последовательности. Компьютеры стали необходимыми в молекулярной биологии, когда белковые последовательности стали доступны после того, как Фредерик Сенгер определил последовательность инсулина в начале 1950-х. Сравнение нескольких последовательностей вручную оказалось непрактичным. Пионером в этой области была Маргарет Окли Дэйхоф (Margaret Oakley Dayhoff). Дэвид Липман (директор Национального центра биотехнологической информации) назвал её «матерью и отцом биоинформатики». Дэйхоф составила одну из первых баз последовательностей белков, первоначально опубликовав в виде книг и стала первооткрывателем методов выравнивания последовательностей и молекулярной эволюции.
Геномы. Поскольку полные последовательности генома стали доступны, снова с новаторской работой Фредерика Сенгера термин «биоинформатика» был переоткрыт и обозначал создание и техническое обслуживание баз данных для хранения биологической информации, такой как последовательности нуклеотидов (база данных GenBank в 1982). Создание таких баз данных включало в себя не только вопросы оформления, но и создание комплексного интерфейса, позволяющего исследователям запрашивать имеющиеся данные и добавлять новые. С публичной доступностью данных, инструменты для их обработки были быстро разработаны и описаны в таких журналах, как «Исследование Нуклеиновых Кислот», который опубликовал специализированные вопросы по инструментам биоинформатики уже в 1982 году.
Цели[править | править код]
Главная цель биоинформатики — способствовать пониманию биологических процессов. Отличие биоинформатики от других подходов состоит в том, что она фокусируется на создании и применении интенсивных вычислительных методов для достижения этой цели. Примеры подобных методов: распознавание образов, data mining, алгоритмы машинного обучения и визуализация биологических данных. Основные усилия исследователей направлены на решение задач выравнивания последовательностей, нахождения генов (поиск региона ДНК, кодирующего гены), расшифровки генома, конструирования лекарств, разработки лекарств, выравнивания структуры белка, предсказания структуры белка, предсказания экспрессии генов и взаимодействий «белок-белок», полногеномного поиска ассоциаций и моделирования эволюции.
Биоинформатика сегодня подразумевает создание и совершенствование баз данных, алгоритмов, вычислительных и статистических методов и теории для решения практических и теоретических проблем, возникающих при управлении и анализе биологических данных.
Анализ генетических последовательностей[править | править код]
Обработка гигантского количества данных, получаемых при секвенировании, является одной из важнейших задач биоинформатикиC тех пор как в 1977 году был секвенирован фаг Phi-X174[en], последовательности ДНК всё большего числа организмов были дешифрованы и сохранены в базах данных. Эти данные используются для определения последовательностей белков и регуляторных участков. Сравнение генов в рамках одного или разных видов может продемонстрировать сходство функций белков или отношения между видами (таким образом могут быть составлены Филогенетические деревья). С возрастанием количества данных уже давно стало невозможным вручную анализировать последовательности. В наши дни для поиска по геномам тысяч организмов, состоящих из миллиардов пар нуклеотидов используются компьютерные программы. Программы могут однозначно сопоставить (выровнять) похожие последовательности ДНК в геномах разных видов; часто такие последовательности несут сходные функции, а различия возникают в результате мелких мутаций, таких как замены отдельных нуклеотидов, вставки нуклеотидов, и их «выпадения» (делеции). Один из вариантов такого выравнивания применяется при самом процессе секвенирования. Так называемая техника «дробного секвенирования» (которая была, например, использована Институтом Генетических Исследований [en] для секвенирования первого бактериального генома, Haemophilus influenzae) вместо полной последовательности нуклеотидов даёт последовательности коротких фрагментов ДНК (каждый длиной около 600—800 нуклеотидов). Концы фрагментов накладываются друг на друга и, совмещённые должным образом, дают полный геном. Такой метод быстро даёт результаты секвенирования, но сборка фрагментов может быть довольно сложной задачей для больших геномов. В проекте по расшифровке генома человека сборка заняла несколько месяцев компьютерного времени. Сейчас этот метод применяется для практически всех геномов, и алгоритмы сборки геномов являются одной из острейших проблем биоинформатики на сегодняшний момент.
Другим примером применения компьютерного анализа последовательностей является автоматический поиск генов и регуляторных последовательностей в геноме. Не все нуклеотиды в геноме используются для задания последовательностей белков. Например, в геномах высших организмов, большие сегменты ДНК явно не кодируют белки и их функциональная роль неизвестна. Разработка алгоритмов выявления кодирующих белки участков генома является важной задачей современной биоинформатики.
Биоинформатика помогает связать геномные и протеомные проекты, к примеру, помогая в использовании последовательности ДНК для идентификации белков.
Аннотация геномов[править | править код]
В контексте геномики аннотация — процесс маркировки генов и других объектов в последовательности ДНК. Первая программная система аннотации геномов была создана в 1995 году Оуэном Уайтом (англ. Owen White), работавшим в команде Института Геномных Исследований (англ. The Institute for Genomic Research), секвенировавшей и проанализировавшей первый декодированный геном свободноживущего организма, бактерии Haemophilus influenzae. Доктор Уайт построил систему для нахождения генов (участок ДНК, задающий последовательность определённого полипептида либо функциональной РНК), тРНК и других объектов ДНК и сделал первые обозначения функций этих генов. Большинство современных систем аннотации генома работают сходным образом, но такие программы доступные для анализа геномной ДНК, как GeneMark, используются для нахождения генов, кодирующих белок в Haemophilus influenzae, постоянно меняются и совершенствуются.
Вычислительная эволюционная биология[править | править код]
Эволюционная биология исследует происхождение и появление видов, также как их развитие с течением времени. Информатика помогает эволюционным биологам в нескольких аспектах:
- изучать эволюцию большого числа организмов, измеряя изменения в их ДНК, а не только в строении или физиологии;
- сравнивать целые геномы (см. BLAST), что позволяет изучать более комплексные эволюционные события, такие как: дупликация генов, горизонтальный перенос генов, и предсказывать бактериальные специализирующие факторы;
- строить компьютерные модели популяций, чтобы предсказать поведение системы во времени;
- отслеживать появление публикаций, содержащих информацию о большом количестве видов.
Область в компьютерных науках, которая использует генетические алгоритмы, часто путают с компьютерной эволюционной биологией, но две эти области не обязательно связаны. Работа в этой области использует специализированное программное обеспечение для улучшения алгоритмов и вычислений и основывается на эволюционных принципах, таких, как репликация, диверсификация через рекомбинацию или мутации, и выживании в естественном отборе.
Оценка биологического разнообразия[править | править код]
Биологическое разнообразие экосистемы может быть определено как полная генетическая совокупность определённой среды, состоящая из всех обитающих видов, была бы это биоплёнка в заброшенной шахте, капля морской воды, горсть земли или вся биосфера планеты Земля. Для сбора видовых имён, описаний, области распространения, генетической информации используются базы данных. Специализированное программное обеспечение применяется для поиска, визуализации и анализа информации, и, что более важно, предоставления её другим людям. Компьютерные симуляторы моделируют такие вещи, как популяционная динамика, или вычисляют общее генетическое здоровье культуры в агрономии. Один из важнейших потенциалов этой области заключается в анализе последовательностей ДНК или полных геномов целых вымирающих видов, позволяя запомнить результаты генетического эксперимента природы в компьютере и возможно использовать вновь в будущем, даже если эти виды полностью вымрут.
Часто из области рассмотрения биоинформатики выпадают методы оценки других компонентов биоразнообразия — таксонов (в первую очередь видов) и экосистем. В настоящее время математические основания биоинформационных методов для таксонов представлены в рамках такого научного направления как фенетика, или численная таксономия. Методы анализа структуры экосистем рассматриваются специалистами таких направлений как системная экология, биоценометрия.
Основные биоинформатические программы[править | править код]
- ACT (Artemis Comparison Tool) — геномный анализ
- Arlequin — анализ популяционно-генетических данных
- Bioconductor — масштабный FLOSS-проект, предоставляющий множество отдельных пакетов для биоинформатических исследований. Написан на R.
- BioEdit — редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- BioNumerics — коммерческий универсальный пакет программ
- BLAST — поиск родственных последовательностей в базе данных нуклеотидных и аминокислотных последовательностей
- Clustal — множественное выравнивание нуклеотидных и аминокислотных последовательностей
- DnaSP — анализ полиморфизма последовательностей ДНК
- FigTree — редактор филогенетических деревьев
- Genepop — популяционно-генетический анализ
- Genetix — популяционно-генетический анализ (программа доступна только на французском языке)
- JalView — редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- MacClade — коммерческая программа для интерактивного эволюционного анализа данных
- MEGA — молекулярно-эволюционный генетический анализ
- Mesquite — программа для сравнительной биологии на языке Java
- Muscle — множественное сравнение нуклеотидных и аминокислотных последовательностей. Более быстрая и точная по сравнению с ClustalW
- PAUP — филогенетический анализ с использованием метода парсимонии (и других методов)
- PHYLIP — пакет филогенетических программ
- Phylo_win — филогенетический анализ. Программа имеет графический интерфейс.
- PopGene — анализ генетического разнообразия популяций
- Populations — популяционно-генетический анализ
- PSI Protein Classifier — обобщение результатов, полученных с помощью программы PSI-BLAST
- Seaview — филогенетический анализ (с графическим интерфейсом)
- Sequin — депонирование последовательностей в GenBank, EMBL, DDBJ
- SPAdes — сборщик бактериальных геномов
- SplitsTree — программа для построения филогенетических деревьев
- T-Coffee — множественное прогрессивное выравнивание нуклеотидных и аминокислотных последовательностей. Более чувствительное, чем в ClustalW/ClustalX.
- UGENE — свободный русскоязычный инструмент, множественное выравнивание нуклеотидных и аминокислотных последовательностей, филогенетический анализ, аннотирование, работа с базами данных.
- Velvet — сборщик геномов
- ZENBU — обобщение результатов
Биоинформатика и вычислительная биология[править | править код]
Под биоинформатикой понимают любое использование компьютеров для обработки биологической информации. На практике, иногда это определение более узкое, под ним понимают использование компьютеров для обработки экспериментальных данных по структуре биологических макромолекул (белков и нуклеиновых кислот) с целью получения биологически значимой информации. В свете изменения шифра научных специальностей (03.00.28 «Биоинформатика» превратилась в 03.01.09 «Математическая биология, биоинформатика») поле термина «биоинформатика» расширилось и включает все реализации математических алгоритмов, связанных с биологическими объектами.
Термины биоинформатика и «вычислительная биология» часто употребляются как синонимы, хотя последний чаще указывает на разработку алгоритмов и конкретные вычислительные методы. Считается, что не всякое использование вычислительных методов в биологии является биоинформатикой, например, математическое моделирование биологических процессов — это не биоинформатика.[источник не указан 3462 дня]
Биоинформатика использует методы прикладной математики, статистики и информатики. Исследования в вычислительной биологии нередко пересекаются с системной биологией. Основные усилия исследователей в этой области направлены на изучение геномов, анализ и предсказание структуры белков, анализ и предсказание взаимодействий молекул белка друг с другом и другими молекулами, а также реконструкция эволюции.
Биоинформатика и её методы используются также в биохимии, биофизике, экологии и в других областях. Основная линия в проектах биоинформатики — это использование математических средств для извлечения полезной информации из «шумных» или слишком объёмных данных о структуре ДНК и белков, полученных экспериментально.
К структурной биоинформатике относится разработка алгоритмов и программ для предсказания пространственной структуры белков. Темы исследований в структурной биоинформатике:
- Рентгеноструктурный анализ (РСА) макромолекул
- Индикаторы качества модели макромолекулы, построенной по данным РСА
- Алгоритмы вычисления поверхности макромолекулы
- Алгоритмы нахождения гидрофобного ядра молекулы белка
- Алгоритмы нахождения структурных доменов белков
- Пространственное выравнивание структур белков
- Структурные классификации доменов SCOP и CATH
- Молекулярная динамика
что нужно знать о биоинформатике / EPAM corporate blog / Habr
Если спросить случайного прохожего, что такое биология, он наверняка ответит что-то вроде «наука о живой природе». Про информатику скажет, что она имеет дело с компьютерами и информацией. Если мы не побоимся быть навязчивыми и зададим ему третий вопрос – что такое биоинформатика? – тут-то он наверняка и растеряется. Логично: про эту область знаний даже в ЕРАМ знает далеко не каждый – хотя в нашей компании и биоинформатики есть. Давайте разбираться, для чего эта наука нужна человечеству вообще и ЕРАМ в частности: в конце концов, вдруг нас на улице об этом спросят.
Почему биология перестала справляться без информатики и при чем тут рак
Чтобы провести исследование, биологам уже недостаточно взять анализы и посмотреть в микроскоп. Современная биология имеет дело с колоссальными объемами данных. Часто обработать их вручную просто невозможно, поэтому многие биологические задачи решаются вычислительными методами. Не будем далеко ходить: молекула ДНК настолько мала, что разглядеть ее под световым микроскопом нельзя. А если и можно (под электронным), всё равно визуальное изучение не помогает решить многих задач.
ДНК человека состоит из трех миллиардов нуклеотидов – чтобы вручную проанализировать их все и найти нужный участок, не хватит и целой жизни. Ну, может и хватит – одной жизни на анализ одной молекулы – но это слишком долго, дорого и малопродуктивно, так что геном анализируют при помощи компьютеров и вычислений.
Биоинформатика — это и есть весь набор компьютерных методов для анализа биологических данных: прочитанных структур ДНК и белков, микрофотографий, сигналов, баз данных с результатами экспериментов и т. д.

Иногда секвенировать ДНК нужно, чтобы подобрать правильное лечение. Одно и то же заболевание, вызванное разными наследственными нарушениями или воздействием среды, нужно лечить по-разному. А еще в геноме есть участки, которые не связаны с развитием болезни, но, например, отвечают за реакцию на определенные виды терапии и лекарств. Поэтому разные люди с одним и тем же заболеванием могут по-разному реагировать на одинаковое лечение.
Еще биоинформатика нужна, чтобы разрабатывать новые лекарства. Их молекулы должны иметь определенную структуру и связываться с определенным белком или участком ДНК. Смоделировать структуру такой молекулы помогают вычислительные методы.
Достижения биоинформатики широко применяют в медицине, в первую очередь в терапии рака. В ДНК зашифрована информация о предрасположенности и к другим заболеваниям, но над лечением рака работают больше всего. Это направление считается самым перспективным, финансово привлекательным, важным – и самым сложным.
Биоинформатика в ЕРАМ
В ЕРАМ биоинформатикой занимается подразделение Life Sciences. Там разрабатывают программное обеспечение для фармкомпаний, биологических и биотехнологических лабораторий всех масштабов — от стартапов до ведущих мировых компаний. Справиться с такой задачей могут только люди, которые разбираются в биологии, умеют составлять алгоритмы и программировать.
Биоинформатики – гибридные специалисты. Сложно сказать, какое знание для них первично: биология или информатика. Если так ставить вопрос, им нужно знать и то и другое. В первую очередь важны, пожалуй, аналитический склад ума и готовность много учиться. В ЕРАМ есть и биологи, которые доучились информатике, и программисты с математиками, которые дополнительно изучали биологию.
Как становятся биоинформатиками
Мария Зуева, разработчик:
«Я получила стандартное ИТ-образование, потом училась на курсах ЕРАМ Java Lab, где увлеклась машинным обучением и Data Science. Когда я выпускалась из лаборатории, мне сказали: «Сходи в Life Sciences, там занимаются биоинформатикой и как раз набирают людей». Не лукавлю: тогда я услышала слово «биоинформатика» в первый раз. Прочитала про нее на Википедии и пошла.
Тогда в подразделение набрали целую группу новичков, и мы вместе изучали биоинформатику. Начали с повторения школьной программы про ДНК и РНК, затем подробно разбирали существующие в биоинформатике задачи, подходы к их решению и алгоритмы, учились работать со специализированным софтом».
Геннадий Захаров, бизнес-аналитик:
«По образованию я биофизик, в 2012-м защитил кандидатскую по генетике. Какое-то время работал в науке, занимался исследованиями – и продолжаю до сих пор. Когда появилась возможность применить научные знания в производстве, я тут же за нее ухватился.
Для бизнес-аналитика у меня весьма специфическая работа. Например, финансовые вопросы проходят мимо меня, я скорее эксперт по предметной области. Я должен понять, чего от нас хотят заказчики, разобраться в проблеме и составить высокоуровневую документацию – задание для программистов, иногда сделать работающий прототип программы. По ходу проекта я поддерживаю контакт с разработчиками и заказчиками, чтобы те и другие были уверены: команда делает то, что от нее требуется. Фактически я переводчик с языка заказчиков – биологов и биоинформатиков – на язык разработчиков и обратно».
Как читают геном
Чтобы понять суть биоинформатических проектов ЕРАМ, сначала нужно разобраться, как секвенируют геном. Дело в том, что проекты, о которых мы будем говорить, напрямую связаны с чтением генома. Обратимся за объяснением к биоинформатикам.
Михаил Альперович, глава юнита биоинформатики:
«Представьте, что у вас есть десять тысяч экземпляров «Войны и мира». Вы пропустили их через шредер, хорошенько перемешали, наугад вытащили из этой кучи ворох бумажных полосок и пытаетесь собрать из них исходный текст. Вдобавок у вас есть рукопись «Войны и мира». Текст, который вы соберете, нужно будет сравнить с ней, чтобы отловить опечатки (а они обязательно будут). Примерно так же читают ДНК современные машины-секвенаторы. ДНК выделяют из клеточных ядер и делят на фрагменты по 300–500 пар нуклеотидов (мы помним, что в ДНК нуклеотиды связаны друг с другом попарно). Молекулы дробят, потому что ни одна современная машина не может прочитать геном от начала до конца. Последовательность слишком длинная, и по мере ее прочтения накапливаются ошибки.
Вспоминаем «Войну и мир» после шредера. Чтобы восстановить исходный текст романа, нам нужно прочитать и расположить в правильном порядке все кусочки романа. Получается, что мы читаем книгу несколько раз по крошечным фрагментам. То же с ДНК: каждый участок последовательности секвенатор прочитывает с многократным перекрытием – ведь мы анализируем не одну, а множество молекул ДНК.
Полученные фрагменты выравнивают – «прикладывают» каждый из них к эталонному геному и пытаются понять, какому участку эталона соответствует прочитанный фрагмент. Затем в выравненных фрагментах находят вариации – значащие отличия прочтений от эталонного генома (опечатки в книге по сравнению с эталонной рукописью). Этим занимаются программы – вариант-коллеры (от англ. variant caller – выявитель мутаций). Это самая сложная часть анализа, поэтому различных программ – вариант-коллеров много и их постоянно совершенствуют и разрабатывают новые.
Подавляющее большинство найденных мутаций нейтральны и ни на что не влияют. Но есть и такие, в которых зашифрованы предрасположенность к наследственным заболеваниям или способность откликаться на разные виды терапии».

Для анализа берут образец, в котором находится много клеток — а значит, и копий полного набора ДНК клетки. Каждый маленький фрагмент ДНК прочитывают несколько раз, чтобы минимизировать вероятность ошибки. Если пропустить хотя бы одну значащую мутацию, можно поставить пациенту неверный диагноз или назначить неподходящее лечение. Прочитать каждый фрагмент ДНК по одному разу слишком мало: единственное прочтение может быть неправильным, и мы об этом не узнаем. Если мы прочитаем тот же фрагмент дважды и получим один верный и один неверный результат, нам будет сложно понять, какое из прочтений правдивое. А если у нас сто прочтений и в 95 из них мы видим один и тот же результат, мы понимаем, что он и есть верный.
Геннадий Захаров:
«Для анализа раковых заболеваний секвенировать нужно и здоровую, и больную клетку. Рак появляется в результате мутаций, которые клетка накапливает в течение своей жизни. Если в клетке испортились механизмы, отвечающие за ее рост и деление, то клетка начинает неограниченно делиться вне зависимости от потребностей организма, т. е. становится раковой опухолью. Чтобы понять, чем именно вызван рак, у пациента берут образец здоровой ткани и раковой опухоли. Оба образца секвенируют, сопоставляют результаты и находят, чем один отличается от другого: какой молекулярный механизм сломался в раковой клетке. Исходя из этого подбирают лекарство, которое эффективно против клеток с “поломкой”».
Биоинформатика: производство и опенсорс
У подразделения биоинформатики в ЕРАМ есть и производственные, и опенсорс-проекты. Причем часть производственного проекта может перерасти в опенсорс, а опенсорсный проект – стать частью производства (например, когда продукт ЕРАМ с открытым кодом нужно интегрировать в инфраструктуру клиента).
Проект №1: вариант-коллер
Для одного из клиентов – крупной фармацевтической компании – ЕРАМ модернизировал программу вариант-коллер. Ее особенность в том, что она способна находить мутации, недоступные другим аналогичным программам. Изначально программа была написана на языке Perl и обладала сложной логикой. В ЕРАМ программу переписали на Java и оптимизировали – теперь она работает в 20, если не в 30 раз быстрее.
Исходный код программы доступен на GitHub.
Проект №2: 3D-просмотрщик молекул
Для визуализации структуры молекул в 3D есть много десктоп- и веб-приложений. Представлять, как молекула выглядит в пространстве, крайне важно, например, для разработки лекарств. Предположим, нам нужно синтезировать лекарство, обладающее направленным действием. Сначала нам потребуется спроектировать молекулу этого лекарства и убедиться, что она будет взаимодействовать с нужными белками именно так, как нужно. В жизни молекулы трехмерные, поэтому анализируют их тоже в виде трехмерных структур.
Для 3D-просмотра молекул ЕРАМ сделал онлайн-инструмент, который изначально работал только в окне браузера. Потом на основании этого инструмента разработали версию, которая позволяет визуализировать молекулы в очках виртуальной реальности HTC Vive. К очкам прилагаются контроллеры, которыми молекулу можно поворачивать, перемещать, подставлять к другой молекуле, поворачивать отдельные части молекулы. Делать всё это в 3D куда удобнее, чем на плоском мониторе. Эту часть проекта биоинформатики ЕРАМ делали совместно с подразделением Virtual Reality, Augmented Reality and Game Experience Delivery.
Программа только готовится к публикации на GitHub, зато пока есть ссылка, по которой можно посмотреть ее демо-версию.
Как выглядит работа с приложением, можно узнать из видео.
Проект №3: геномный браузер NGB
Геномный браузер визуализирует отдельные прочтения ДНК, вариации и другую информацию, сгенерированную утилитами для анализа генома. Когда прочтения сопоставлены с эталонным геномом и мутации найдены, ученому остается проконтролировать, правильно ли сработали машины и алгоритмы. От того, насколько точно выявлены мутации в геноме, зависит, какой диагноз поставят пациенту или какое лечение ему назначат. Поэтому в клинической диагностике контролировать работу машин должен ученый, а помогает ему в этом геномный браузер.
Биоинформатикам-разработчикам геномный браузер помогает анализировать сложные случаи, чтобы найти ошибки в работе алгоритмов и понять, как их можно улучшить.
Новый геномный браузер NGB (New Genome Browser) от ЕРАМ работает в вебе, но по скорости и функционалу не уступает десктопным аналогам. Это продукт, которого не хватало на рынке: предыдущие онлайновые инструменты работали медленнее и умели делать меньше, чем десктопные. Сейчас многие клиенты выбирают веб-приложения из соображений безопасности. Онлайн-инструмент позволяет ничего не устанавливать на рабочий компьютер ученого. С ним можно работать из любой точки мира, зайдя на корпоративный портал. Ученому не обязательно всюду возить за собой рабочий компьютер и скачивать на него все необходимые данные, которых может быть очень много.
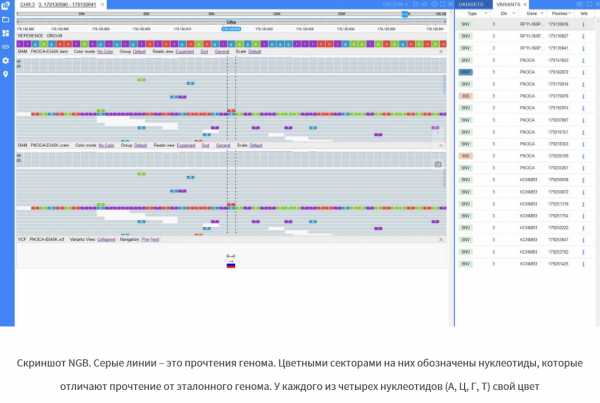
Геннадий Захаров, бизнес-аналитик:
«Над опенсорсными утилитами я работал частично как заказчик: ставил задачу. Я изучал лучшие решения на рынке, анализировал их преимущества и недостатки, искал, как можно их усовершенствовать. Нам нужно было сделать веб-решения не хуже десктопных аналогов и при этом добавить в них что-то уникальное.
В 3D-просмотрщике молекул это была работа с виртуальной реальностью, а в геномном браузере – улучшенная работа с вариациями. Мутации бывают сложными. Перестройки в раковых клетках иногда затрагивают огромные области. В них появляются лишние хромосомы, куски хромосом и целые хромосомы исчезают или объединяются в случайном порядке. Отдельные куски генома могут копироваться по 10–20 раз. Такие данные, во-первых, сложнее получить из прочтений, а во-вторых, сложнее визуализировать.
Мы разработали визуализатор, который правильно читает информацию о таких протяженных структурных перестройках. Еще мы сделали набор визуализаций, который при контакте хромосом показывает, образовались ли из-за этого контакта гибридные белки. Если протяженная вариация затрагивает несколько белков, мы по клику можем рассчитать и показать, что происходит в результате такой вариации, какие гибридные белки получаются. В других визуализаторах ученым приходилось отслеживать эту информацию вручную, а в NGB – в один клик».
Как изучать биоинформатику
Мы уже говорили, что биоинформатики – гибридные специалисты, которые должны знать и биологию, и информатику. Самообразование играет в этом не последнюю роль. Конечно, в ЕРАМ есть вводный курс в биоинформатику, но рассчитан он на сотрудников, которым эти знания пригодятся на проекте. Занятия проводятся только в Санкт-Петербурге. И всё же, если биоинформатика вам интересна, возможность учиться есть:
1) Вводный курс в генетическую диагностику от компании 23andme.
2) Несколько курсов на Coursera (в том числе пара курсов на русском: введение в биоинформатику и в метагеномику).
3) Курсы на Stepik от института биоинформатики: молекулярная биология и генетика, молекулярная филогенетика, генная инженерия и введение в технологии высокоэффективного секвенирования. Полный список курсов от института можно посмотреть на его официальном сайте.
4) Лекции Павла Певзнера – профессора Калифорнийского университета в Сан-Диего, специалиста в области биоинформатики.
5) Если вы живете в Санкт-Петербурге, можно прийти на гостевые лекции в институт биоинформатики – это бесплатно.
где учиться, зарплата, плюсы и минусы
Биоинформатик – человек, занимающийся анализом медико-биологических данных. Он разрабатывает, а также применяет алгоритмические, вычислительные и иные методы, позволяющие узнать больше об информации, заключенной в наших клетках, иных биологических данных. Профессия подходит тем, кого интересует физика, математика, химия и биология (см. выбор профессии по интересу к школьным предметам).
Читайте также:
Краткое описание
Современные методы диагностики и исследований приводят к росту количества научных данных, которые вручную обрабатывать очень сложно. В этом случае на помощь приходит биоинформатика, которая как междисциплинарная область науки сформировалась во второй половине XX века. Биоинформатики пользуются элементами прикладной математики, статистики, а также информатики. Во время работы они оперируют следующими знаниями:
- языки программирования, преимущественно Java, С, С++, С#, R;
- язык разметки HTML;
- программы: ACT, BLAST, Clustal и иные;
- SQL, CUDA.
Рассмотрим основные области исследования:
- анализ генетических последовательностей, эволюционная вычислительная биология;
- проведение оценки биологического разнообразия, аннотация геномов.
Профессия молодая, в дальнейшем она будет развиваться еще более стремительно, ведь применение вычислительных методов гарантирует высокую точность, скорость и исключает человеческий фактор. Технологии биоинформатики необходимы в биохимии, биофизике, экологии, фармакологии, сельском хозяйстве, генетике и других сферах.
Особенности профессии
Биоинформатика находится на стыке медицины, биологии, прикладной математики, информатики. В обязанности людей, выбравших это направления, входит решение глобальных задач:
- поиск методов лечения онкологических, хронических, аутоиммунных заболеваний;
- продление срока жизни населения, улучшение экологической ситуации, поиск генома долголетия;
- разработка, планирование, внедрение математических методов, алгоритмов, программ, используемых для а
анализ данных, нейросети, и их применение в биологии и медицине / Habr
Почти год назад, летом 2017 года, на базе МФТИ состоялась традиционная летняя школа от Института биоинформатики. Основной темой школы в этом году стал интеллектуальный анализ данных. Почему? Количество получаемых данных в биологии и медицине растет с невероятной скоростью. В то же время обнаружить ранее неизвестные вещи в таком объеме информации вручную физически невозможно (да и классическими алгоритмами уже тоже сложновато), поэтому приходится использовать статистику и дополнять естественный интеллект искусственным.Именно этим активно и занимались участники летней школы. В этом посте собрано 22 видеозаписи лекций со слайдами и описанием для всех интересующихся темой анализа данных в биоинформатике. Лекции, которые можно смотреть без дополнительной подготовки, отмечены звёздочкой «*» (таких половина).

1*. Введение в биоинформатику (Александр Предеус, Институт биоинформатики)
Видео | Слайды
В лекции рассмотрены основные области, в которых работают биоинформатики в науке и индустрии, особенности биоинформатики и причины ее популярности сегодня.

2*. Введение в машинное обучение (Григорий Сапунов, Intento)
Видео | Слайды
Постоянный рост количества данных способствует развитию все более и более сложных процессов обработки, поиска и извлечения информации. Один из способов решения подобных задач заключается в использовании искусственного интеллекта. Эта лекция посвящена краткому введению в основы машинного обучения. Григорий рассказал общую терминологию в этой области, а также описал виды задач, решаемых машинным обучением. Помимо этого, лекция знакомит с основными этапами машинного обучения, видами моделей и метриками качества полученных данных.
3*. Введение в Deep Learning (Григорий Сапунов, Intento)
Видео | Слайды
Глубокое обучение (или deep learning) в настоящее время набирает популярность из-за возможности не прописывать конкретные алгоритмы для решения задачи, а использовать обучение представлениям. Развитию этих методов также способствует увеличение вычислительной мощности процессоров. Лекция посвящена основам нейросетей: их видам (полносвязные нейросети, автоэнкодеры, свёрточные, рекуррентные) и решаемым ими задачам. Отдельно Григорий обрисовал современное состояние и тренды.

4*. Введение в онкогеномику и анализ омиксных данных в онкологии (Михаил Пятницкий, НИИ биомедицинской химии им. В.Н.Ореховича)
Видео | Слайды
Секвенирование человеческого генома, изучение человеческих генетических вариаций, секвенирование метагенома человека, транскриптомный анализ человеческих тканей — все эти биологические методы в приложении к “Big Data” дали ученым большой объем ценной информации о том, что отличает человека от других животных. Эта лекция посвящена «омикам» и их практическому использованию. Отдельно Михаил затронул использование этих данных в онкологии.

5. Мультиомика в биологии: интеграция технологий (Константин Оконечников, German Cancer Research Center)
Видео | Слайды
Бурное развитие экспериментальных технологий в молекулярной биологии, таких как например, секвенирование, позволили совместить в себе изучение большого спектра функциональных процессов происходящих в клетках, органах или даже целом организме. В лекции рассмотрено как правильно совмещать массивные экспериментальные данные, полученные из геномики, транксриптомики и эпигеномики для установления связей между компонентами происходящих биологических процессов. Наглядные примеры применения мультиомики выбраны из высоко востребованной области исследований раковых заболеваний с фокусом на педиатрическую онкологию.

6. Количественная генетика: история и перспективы (Юрий Аульченко, лаборатория теоретической и прикладной функциональной геномики ФЕН НГУ, группа методов генетического анализа, ИЦиГ СО РАН)
Видео | Слайды
Количественная генетика — точная наука, которая основывается на небольшом числе ключевых наблюдений и базовых моделей, позволяющих дать количественное описание природных (микро)эволюционных явлений и предсказать результаты генетических экспериментов. Она использует мощный математический аппарат. Многие современные методы статистики были изначально разработаны для решения проблем количественной генетики. Прорывное развитие молекулярно-биологических технологий за последнее десятилетие позволило характеризовать сотни тысяч живых организмов по миллионам геномных и других «омиксных» параметров. Общее количество проведенных экспериментов и уже накопленных данных колоссально. Актуальная задача современной количественной генетики — разработка моделей, которые позволят описать наследования многоуровневых фенотипических высокой размерности. В своей лекции Юрий дал краткий обзор истории количественной генетики и проблем, которые стоят перед этой наукой.

7*. Технологии секвенирования (Кирилл Григорьев, Caribbean Genome Center, University of Puerto Rico)
Видео | Слайды
Развитие и эволюция процессов секвенирования неразрывно связаны с эволюцией технологических возможностей. Лекция показывает историю и процесс развития технологий секвенирования от Сэнгера до наших дней. Отдельно Кирилл рассказал про преимущества и недостатки каждого из существующих в настоящее время методов, а также о характере получаемых данных и их применении в различных областях.
8. Транскриптомика: практические методы и применяемые алгоритмы (Александр Предеус, Институт биоинформатики)
Видео | Слайды
Транскриптомика уверенно заняла место в списке самых популярных задач, встающих перед NGS-биоинформатиками. Дифференциальный анализ экспрессии генов, кластеризация экспрессионных данных, и интерпретация полученных данных в терминах метаболических и сигнальных каскадов позволяют получить богатейшую информацию о практически любой системе. В лекции рассмотрены лучшие пайплайны, основные проблемные места в дизайне экспериментов и обработке, а также практические случаи удачного применения транскриптомных подходов.

9. Анализ данных NGS в медицинской генетике: определение, аннотация и интерпретация генетических вариантов (Юрий Барбитов, СПбГУ, Александр Предеус, Институт биоинформатики)
Видео | Слайды
Использование секвенирования нового поколения давно ушло за пределы классической науки и успешно применяется во многих других областях, в том числе в здравоохранении. Лекция посвящена ключевым аспектам анализа данных секвенирования нового поколения в медицинской генетике. Юрий показал весь путь от получения сырых ридов до постановки диагноза, с упоминанием трудностей, возникающих при определении, аннотации и интерпретации генетических вариантов. Отдельно он затронул распространенные ошибки, допускаемые на каждом из этапов обработки данных. В заключение дан краткий обзор перспективных направлений исследований, способных улучшить точность постановки диагноза с использованием методов высокопроизводительного секвенирования
10. Практическое применение ChIP-Seq и родственных методов (Александр Предеус, Институт биоинформатики)
Видео | Слайды
Методы ChIP-Seq, а также «геномного футпринтинга» (ATAC-Seq, FAIRE-Seq, DNase-Seq) широко применяются для нахождения механизмов регуляции биологических процессов, в частности, для транскрипционной регуляции. Потенциальное пространство изучаемых факторов очень многомерно, однако селективный подход позволяет получить богатую информацию о регуляции в системе на основании всего нескольких экспериментов. На примере конфликтующих современных теорий, Александр показал основные сложности интерпретации регуляторной информации, и способы консолидации полученных результатов.

11*. Что можно делать с данными iScan (Татьяна Татаринова, University of La Verne )
Видео | Слайды
Компания Illumina выпускает большое количество приборов под различные нужды. Чипирование позволяет быстро обнаруживать однонуклеотидные полиморфизмы (SNP) для большого количества образцов. Лекция посвящена обзору данных чипов iScan и их применению в клинической диагностике.

12. Глубокое обучение в вычислительной биологии (Дмитрий Фишман, University of Tartu)
Видео | Слайды
Глубокое обучение активно используется не только для улучшения машинного перевода или распознавания речи, но и позволяет решить многие проблемы в области вычислительной биологии. Лекция посвящена применению методов глубокого обучения на конкретных биологических примерах. Дмитрий рассказал о том, что нового происходит в биологии и медицине с использованием глубокого обучения, и можно ли говорить о том, что машины революционизируют медицину и биологию.

13*. Применение методов машинного обучения для поиска потенциальных патогенных мутаций в геноме человека (Анна Ершова, МФТИ, НИИ физико-химической биологии МГУ им. М.В. Ломоносова, ФНИЦ эпидемиологии и микробиологии им. Н.Ф. Гамалеи)
Видео | Слайды
Поиск патогенных мутаций стал актуальным в связи с секвенированием генома человека. Однако, вручную такую задачу решить просто невозможно. Лекция посвящена тому, как машинное обучение может помочь справиться с этой задачей.

14*. Иммуноинформатика (Вадим Назаров, НИУ ВШЭ, ИБХ РАН)
Видео | Слайды
Машинное обучение уже довольно давно активно применяется в самых разных сферах жизни, но в иммунологии для него нашли место совсем недавно. В этой лекции Вадим рассказал о нескольких примерах применения машинного и глубинного обучения в иммунологии, включая задачу предсказания связывания МНС-пептид комплексов и анализа репертуаров Т-клеточных рецепторов.

15*. Изучение адаптации к хозяину и развития резистентности в вирусах ВИЧ и гепатита С с помощью методов структурной биоинформатики (Ольга Калинина, Институт информатики общества Макса Планка)
Видео | Слайды
Вирус иммунодефицита человека (ВИЧ) и вирус гепатита С вызывают тяжелые заболевания, которые с трудом поддаются терапии. Как и многие другие ретро- и РНК-вирусы, эти вирусы быстро эволюционируют и, таким образом, могут приспосабливаться как к воздействию специфических антивирусных препаратов, так и к адаптивному иммунному ответу со стороны организма хозяина. В этой лекции Ольга показала, как с помощью комбинирования анализа последовательностей вирусных белков с анализом их пространственной структуры можно делать предсказания о развитии механизмов резистентности и взаимодействии вирусов с иммунной системой хозяина.

16. Предсказание эффекта мутаций (Василий Раменский, МФТИ)
Видео | Слайды
Современные методы секвенирования дают огромный объем информации о полиморфизме генома, то есть отличиях индивидуальных геномов друг от друга. Эти отличия (варианты) возникают в результате мутаций при репликации ДНК и частично фиксируются в популяции. Распространенность, локализация и функциональный эффект геномных вариантов сильно различаются – от полной летальности до отсутствия какого-либо влияния на индивидуальный фенотип. В лекции рассмотрены современные подходы к предсказанию функционального эффекта вариантов, используемые в персонализированной медицине, медицинской и популяционной генетике.

17. Многомасштабное моделирование и дизайн биологических молекул (Николай Дохолян, University of North Carolina at Chapel Hill)
Видео
Жизнь биологических молекул охватывает масштабы времени и длины, соответствующие шкалам времени и длины от атомного до клеточного. Следовательно, новые подходы к молекулярному моделированию должны быть по своей сути многомасштабными. В своей лекции Николай описал несколько методологий, разработанных в его лаборатории: алгоритм быстрого дискретного молекулярного динамического моделирования, белковый дизайн и инструменты структурной доработки. Используя эти методологии, можно описать несколько приложений, которые проливают свет на молекулярную этиологию кистозного фиброза и находят новые фармацевтические стратегии для борьбы с этим заболеванием, моделируют структуру трехмерной РНК и разрабатывают новые подходы к контролю белков в живых клетках и организмах.

18. Гомологичный фолдинг белков (Павел Яковлев, BIOCAD)
Видео
В современной структурной биологии есть ряд вычислительных методов, позволяющих с высокой достоверностью характеризовать биологические молекулы, их схожесть и различия, способы взаимодействия и функции. Для построения подобных вычислений входным параметром всегда выступает пространственная структура белка, однако ее получение может быть затруднен, несмотря на полувековой прогресс в области кристаллографии. Лекция посвящена решению этой проблемы с помощью гомологичного моделирования структур белков — построения трехмерных структур из схожих фрагментов. Для примера рассмотрены вариабельные домены антител — белков, обладающих уникальным структурным разнообразием вариабельных петель.

19. Как перестать медитировать и начать моделировать (Артур Залевский, МГУ им. М. В. Ломоносова)
Видео | Слайды
Большое количество данных, получаемых методом NGS, позволяет не только получать из этого биологические выводы, но и использовать их для моделирования. Построенные модели позволяют лучше понять биологические данные и получить еще больше биологического смысла из эксперимента. Лекция посвящена моделированию и начальным этапам этого процесса.

20*. Стоя на плечах гигантов, или зачем нужны консорциумы (Герман Демидов, Centre for Genomic Regulation, The Barcelona Institute of Science and Technology, Universitat Pompeu Fabra)
Видео | Слайды
За последние десятилетия развитие биологии было связано с накоплением массивов данных, огромных настолько, что отдельные исследовательские группы уже не справлялись с их биоинформатическим анализом. С целью решить эту проблему начали создаваться консорциумы из десятков лабораторий, такие как Human Genome Project, 1000GP, ENCODE и другие. Благодаря таким коллаборациям, в открытом доступе есть данные разнообразных типов, полученные с помощью различных технологий. Как результат, сравнение новых экспериментальных данных с уже существующими стало стандартной частью любого исследования. Консорциумы производят не только данные, но и биоинформатические пайплайны для их обработки, и стандартные форматы, и процедуры оценки качества. На этой лекции обсуждается, как работают консорциумы, как пользоваться результатами их работы и что делать, если вы вдруг обнаружили себя членом такого консорциума и вам нужно обрабатывать терабайты данных, а потом обмениваться результатами со всеми остальными участниками.

21*. Обзор биоинформатических компаний в России и мире (Андрей Афанасьев, yRisk)
Видео | Слайды
В современном мире наука и бизнес все более и более переплетаются. Не обошел этот тренд и область биоинформатики. Андрей рассказал об ожиданиях и реальности рынка, об историях успеха и историях провалов, о людях и местах, связанных с биоинформатикой.

22. Продвинутый анализ вариаций (SNV, InDel, SV) с помощью геномного браузера NGB (Геннадий Захаров, EPAM, Институт Физиологии им. И.П. Павлова, РАН)
Видео | Слайды
Лекция охватывает процесс визуального анализа простых (SNV, InDel) и структурных вариаций в геномном браузере. Все примеры демонстрируются с использованием браузера NGB, отвечающего большинству требований и рекомендаций анализа структурных вариаций, в том числе различные виды визуализаций и получение аннотаций из внешних баз данных. В лекции на реальных примерах показаны сценарии валидации и анализа последствий простых и структурных вариаций.
Послесловие
Для тех, кто
В 2017 году школа проводилась при поддержке наших постоянных партнеров – компаний JetBrains, BIOCAD и EPAM Systems, за что им огромное спасибо.
Кстати, пост с лекциями позапрошлых школ.
Всем биоинформатики!

что это за наука и зачем она нужна?
Если спросить случайного прохожего, что такое биология, он наверняка ответит что-то вроде «наука о живой природе». Про информатику скажет, что она имеет дело с компьютерами и информацией. Если мы не побоимся быть навязчивыми и зададим ему третий вопрос – что такое биоинформатика? – тут-то он наверняка и растеряется. Логично: это сравнительно новая область знаний, и в школе ее точно не преподают. Давайте разбираться, для чего биоинформатика нужна человечеству: в конце концов, вдруг нас на улице об этом спросят.
Почему биология перестала справляться без информатики и при чем тут рак
Чтобы провести исследование, биологам уже недостаточно взять анализы и посмотреть в микроскоп. Современная биология имеет дело с колоссальными объемами данных. Часто обработать их вручную просто невозможно, поэтому многие биологические задачи решаются вычислительными методами. Не будем далеко ходить: молекула ДНК настолько мала, что разглядеть ее под световым микроскопом нельзя. А если и можно (под электронным), всё равно визуальное изучение не поможет решить всех задач.
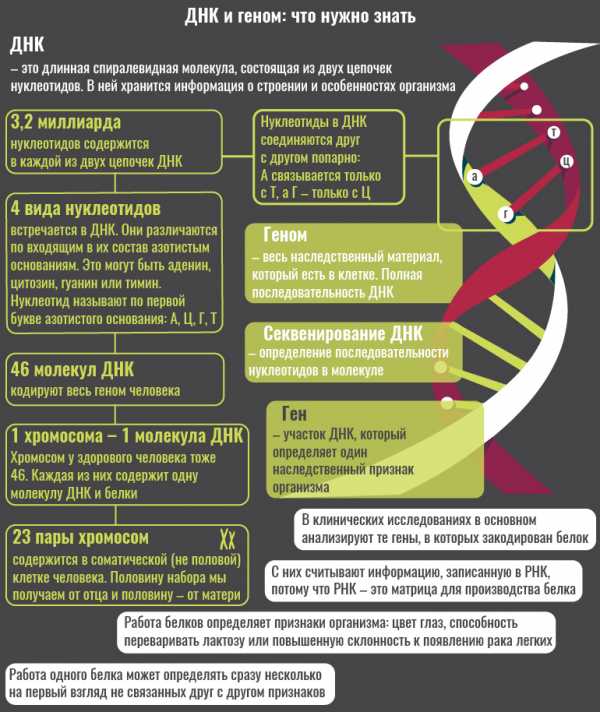
ДНК человека состоит из трех миллиардов нуклеотидов – чтобы вручную проанализировать их все и найти нужный участок, не хватит и целой жизни. Ну, может и хватит – одной жизни на анализ одной молекулы – но это слишком долго, дорого и малопродуктивно, так что геном анализируют при помощи компьютеров и вычислений.
Биоинформатика — это и есть весь набор компьютерных методов для анализа биологических данных: прочитанных структур ДНК и белков, микрофотографий, сигналов, баз данных с результатами экспериментов и т. д.
Иногда секвенировать ДНК нужно, чтобы подобрать правильное лечение. Одно и то же заболевание, вызванное разными наследственными нарушениями или воздействием среды, надо лечить по-разному. А еще в геноме есть участки, которые не связаны с развитием болезни, но, например, отвечают за реакцию на определенные виды терапии и лекарств. Поэтому разные люди с одним и тем же заболеванием могут по-разному реагировать на одинаковое лечение.
Еще биоинформатика нужна, чтобы разрабатывать новые лекарства. Их молекулы должны иметь определенную структуру и связываться с каким-то конкретным белком или участком ДНК. Смоделировать структуру такой молекулы помогают вычислительные методы.
Достижения биоинформатики широко применяют в медицине, в первую очередь в терапии рака. В ДНК зашифрована информация о предрасположенности и к другим заболеваниям, но над лечением рака работают больше всего. Это направление считается самым перспективным, финансово привлекательным, важным – и самым сложным.
Как читают геном
Чтобы понять суть биоинформатических проектов, сначала нужно разобраться, как секвенируют геном.
Представьте, что у вас есть десять тысяч экземпляров «Войны и мира». Вы пропустили их через шредер, хорошенько перемешали, наугад вытащили из этой кучи ворох бумажных полосок и пытаетесь собрать из них исходный текст. Вдобавок у вас есть рукопись «Войны и мира». Текст, который вы соберете, нужно будет сравнить с ней, чтобы отловить опечатки (а они обязательно будут). Примерно так же читают ДНК современные машины-секвенаторы. ДНК выделяют из клеточных ядер и делят на фрагменты по 300–500 пар нуклеотидов (мы помним, что в ДНК нуклеотиды связаны друг с другом попарно). Молекулы дробят, потому что ни одна современная машина не может прочитать геном от начала до конца. Последовательность слишком длинная, и по мере ее прочтения накапливаются ошибки.
Вспоминаем «Войну и мир» после шредера. Чтобы восстановить исходный текст романа, нам нужно прочитать и расположить в правильном порядке все кусочки романа. Получается, что мы читаем книгу несколько раз по крошечным фрагментам. То же с ДНК: каждый участок последовательности секвенатор прочитывает с многократным перекрытием – ведь мы анализируем не одну, а множество молекул ДНК.
Полученные фрагменты выравнивают – «прикладывают» каждый из них к эталонному геному и пытаются понять, какому участку эталона соответствует прочитанный фрагмент. Затем в выровненных фрагментах находят вариации – значащие отличия прочтений от эталонного генома (опечатки в книге по сравнению с эталонной рукописью). Этим занимаются программы – вариант-коллеры (от англ. variant caller – выявитель мутаций). Это самая сложная часть анализа, поэтому различных программ – вариант-коллеров много и их постоянно совершенствуют и разрабатывают новые.
Подавляющее большинство найденных мутаций нейтральны и ни на что не влияют. Но есть и такие, в которых зашифрованы предрасположенность к наследственным заболеваниям или способность откликаться на разные виды терапии при заболеваниях.

Для анализа берут образец, в котором находится много клеток — а значит, и копий полного набора ДНК клетки. Каждый маленький фрагмент ДНК прочитывают несколько раз, чтобы минимизировать вероятность ошибки. Если пропустить хотя бы одну значащую мутацию, можно поставить пациенту неверный диагноз или назначить неподходящее лечение. Прочитать каждый фрагмент ДНК по одному разу слишком мало: единственное прочтение может быть неправильным, и мы об этом не узнаем. Если мы прочитаем тот же фрагмент дважды и получим один верный и один неверный результат, нам будет сложно понять, какое из прочтений правдивое. А если у нас сто прочтений и в 95 из них мы видим один и тот же результат, мы понимаем, что он и есть верный.
Для анализа раковых заболеваний секвенировать нужно и здоровую, и больную клетку. Рак появляется в результате мутаций, которые клетка накапливает в течение своей жизни. Если в клетке испортились механизмы, отвечающие за ее рост и деление, то клетка начинает неограниченно делиться вне зависимости от потребностей организма, т. е. становится раковой опухолью. Чтобы понять, чем именно вызван рак, у пациента берут образец здоровой ткани и раковой опухоли. Оба образца секвенируют, сопоставляют результаты и находят, чем один отличается от другого: какой молекулярный механизм сломался в раковой клетке. Исходя из этого подбирают лекарство, которое эффективно против клеток с «поломкой».
Как изучать биоинформатику
Биоинформатики – гибридные специалисты, которые должны знать и биологию, и информатику. Самообразование играет в этом не последнюю роль. Если биоинформатика вам интересна, вы можете выбрать что-то из вариантов ниже:
- Вводный курс в генетическую диагностику от компании 23andme.
- Несколько курсов на Coursera (в том числе пара курсов на русском: введение в биоинформатику и в метагеномику).
- Курсы на Stepik от Института биоинформатики: молекулярная биология и генетика, молекулярная филогенетика, генная инженерия и введение в технологии высокоэффективного секвенирования. Полный список курсов от института можно посмотреть на его официальном сайте.
- Лекции Павла Певзнера – профессора Калифорнийского университета в Сан-Диего, специалиста в области биоинформатики.
- Если вы живете в Санкт-Петербурге, можно прийти на гостевые лекции в институт биоинформатики – это бесплатно.
взгляд изнутри / Образовательные проекты JetBrains corporate blog / Habr
 Из всех известных мне технических и естественных наук, пожалуй, именно о биоинформатике представление у людей самое плохое. Оно либо в той или иной степени неверное, либо его нет совсем. Когда два года назад я начал заниматься бионформатикой, знаний об этой науке у меня самого не было ровным счетом никаких. Со временем я лучше стал представлять, какие задачи стоят перед биоинформатиками, чем они пользуются, и что может являться результатом их работы.
Из всех известных мне технических и естественных наук, пожалуй, именно о биоинформатике представление у людей самое плохое. Оно либо в той или иной степени неверное, либо его нет совсем. Когда два года назад я начал заниматься бионформатикой, знаний об этой науке у меня самого не было ровным счетом никаких. Со временем я лучше стал представлять, какие задачи стоят перед биоинформатиками, чем они пользуются, и что может являться результатом их работы.У биоинформатиков нет никаких пробирок, реагентов, бактерий, белых халатов. Главные инструменты у них – ноутбук, ручка с бумагой или белая доска с маркером – в общем, всё как у программистов. Да и сама работа очень сильно похожа на работу в IT компании, а лаборатория – на небольшой отдел разработки. А в чем же тогда отличия? Что ж, попробую ответить.
Во-первых, задачи в основном алгоритмические. То есть перед тем как написать программу, надо прочитать несколько статей, подумать самому, обсудить свои идеи с коллегами и только потом приступать к реализации. Во-вторых, работать приходится с большими объемами данных, а поэтому реализация должна быть максимально эффективной. Однако даже эффективная, логичная и идеально отлаженная программа может не дать желаемый результат. Основная причина тому – биологическое происхождение данных, а значит огромное количество ошибок и существенное различие между данными от разных лабораторий.
Еще одно, пожалуй, самое видимое отличие биоинформатики от программирования – это исследования и публикации. Биоинформатика – это наука, а значит просто необходимо быть в курсе всего, что происходит в мире. Для этого и существуют многочисленные конференции, сотрудничества с лабораториями из других стран и, безусловно, публикации – о своих достижениях тоже необходимо рассказывать всем. Без всего этого можно усердно изобретать велосипед.
В общем и целом, впечатление о биоинформатике именно такое, но лучше всего рассказать это на примере, тем более что такой есть, и совсем недалеко. Но обо всем по порядку.
Лаборатория алгоритмической биологии
В 2010-м году в России была запущена программа «мегагрантов». Под руководством ведущих западных ученых (в большинстве случаев давно уехавших из России) стали создаваться новые научные лаборатории. Одной из таких стала лаборатория алгоритмической биологии при СПбАУ под руководством Павла Певзнера – одного из самых известных ученых в своей области. Павел закончил МФТИ, но достаточно скоро после этого уехал в США, занялся Computer Science (а если быть точным, именно биоинформатикой) и сейчас является профессором Университета Калифорнии в Сан-Диего.
Перед тем как рассказать, чем же именно занимаются в лаборатории, стоит ввести читателей в курс дела.
Немного о геномике
Уверен, что каждый читатель слышал слово геном. Для биологов геном – это молекулы ДНК – длинные цепочки, состоящие из четырех нуклеотидов, организованные в хромосомы, свёрнутые в ядре клетки. Мы же видим геном как строку, состоящую опять же из четырех символов (A, C, G, T). Длина генома может достигать миллиардов или даже десятков миллиардов символов. Биологи не умеют считывать геном целиком – только маленькими фрагментами до 150 «букв», да и то с ошибками. Наша задача – восстановить исходный геном по этим кусочкам, или как чаще говорят – собрать.
Для наглядности можно привести такое сравнение: представьте пачку одинаковых газет. Теперь представьте, что эту пачку взрывают и мелкие кусочки бумаги разлетаются, перемешиваются, портятся или даже сгорают целиком. А дальше по куче этого мусора хочется склеить исходную газету.
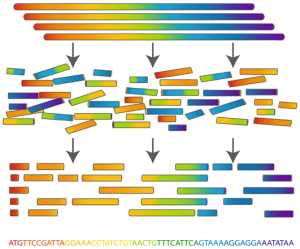 Так же и с геномом. Первые технологии позволяли считывать кусочки генома длиной до нескольких тысяч символов. Эти технологии были невероятно дорогие – на сборку первого человеческого генома были потрачено несколько миллиардов долларов и несколько лет усердной работы сотен сотрудников лабораторий по всему миру. Современные технологии позволяют читать более короткие фрагменты, но на порядок дешевле и в огромном количестве. Обработка гигабайтов входных данных, естественно, производится автоматически. Для этого разрабатываются программы, которые называют геномными сборщиками, или чаще – ассемблерами (от английского assemble). В силу некоторых особенностей исходных геномов (например, повторяющихся регионов), а также большого числа ошибок во входных данных, результатом работы сборщика является не целый геном, а лишь достаточно продолжительные его участки. Чем длиннее полученные участки, чем больше они похожи на исходный геном, тем качественней считается результат.
Так же и с геномом. Первые технологии позволяли считывать кусочки генома длиной до нескольких тысяч символов. Эти технологии были невероятно дорогие – на сборку первого человеческого генома были потрачено несколько миллиардов долларов и несколько лет усердной работы сотен сотрудников лабораторий по всему миру. Современные технологии позволяют читать более короткие фрагменты, но на порядок дешевле и в огромном количестве. Обработка гигабайтов входных данных, естественно, производится автоматически. Для этого разрабатываются программы, которые называют геномными сборщиками, или чаще – ассемблерами (от английского assemble). В силу некоторых особенностей исходных геномов (например, повторяющихся регионов), а также большого числа ошибок во входных данных, результатом работы сборщика является не целый геном, а лишь достаточно продолжительные его участки. Чем длиннее полученные участки, чем больше они похожи на исходный геном, тем качественней считается результат.
Задача сборки генома
Если взять задачу сборки генома в самом общем случае – это будет не что иное, как задача о надстроке (shortest superstring problem), которая формулируется следующим образом: найти кратчайшую строку, такую, что каждая строка из заданного набора являлась бы её подстрокой. Эта задача является NP-полной. Но если предположить, что у нас есть все возможные подстроки исходной строки одинаковой длины, задачу можно решить за полиномиальное время. Сборка генома – это именно такой случай. В 2001 году Павлом Певзнером был предложен эффективный подход сборки геномов с использованием графа де Брюйна. Основная идея этого подхода используется почти в каждом современном геномном ассемблере. Однако на практике все сильно усложняется вышеупомянутыми биологическими ошибками, и поэтому основная задача – разработка эвристик для разного рода подзадач, возникающих при сборке геномов.
В лаборатории алгоритмической биологии было решено сфокусироваться именно на разработке ассемблера. Безусловно, на момент создания лаборатории существовало огромное количество геномных сборщиков. Зачем было тогда создавать еще один? На самом деле, задача сборки оказывается намного более широкой, чем кажется на первый взгляд. Биологи производят огромное количество различных типов входных данных, для каждого из которых требуется разработка новых методов, учитывающих их специфику. Кроме того, сборка генома включает в себя большое число этапов и алгоритмов, поэтому даже несмотря на то, что все современные ассемблеры используют один и тот же подход, их результаты могут очень сильно отличаться. Перед лабораторией ставилась задача получить ассемблер, который по многим параметрам превосходил бы существующие.
Путь в биоинформатику
В биоинформатику я попал, можно сказать, случайно. Я учился в магистратуре СПбАУ и, как и каждому студенту, в начале семестра мне необходимо было выбрать научно-исследовательскую работу. Чтобы попробовать себя в новой области, я выбрал биоинформатический проект. По началу пугало то, что, возможно, придется учить биологию, вместо того чтобы разрабатывать и реализовывать алгоритмы. Однако опасения, к счастью, не оправдались – погружение в эту предметную область происходит точно так же, как и в любую другую. Постепенно начинаешь понимать больше, узнаешь что-то новое, и даже если биология была далеко не самым любимым предметом в школе, интерес к ней появляется достаточно быстро. Почти сразу я понял, что биоинформатика – это именно то, чем я хотел заниматься – программирование с элементами исследовательской работы и интересной предметной областью.
Пока я занимался своим проектом, организовалась лаборатория алгоритмической биологии, о которой я упоминал. Летом 2011 я успешно прошел в ней стажировку и остался как постоянный научный сотрудник. Если говорить о лаборатории в целом – огромное количество различных интересных проектов, которые далеко не ограничиваются сборкой геномов, сотрудничество с западными лабораториями, научные конференции, постоянная возможность узнавать что-нибудь новое и, конечно, очень хороший коллектив.
Можно было бы наверное еще долго говорить и о работе в лаборатории, и о биоинформатике в целом, в которой еще море открытых проблем, и о конкретных подходах и алгоритмах в разных задачах. Но нельзя объять необъятное, а посему рассказ будет о чем-то одном и уже в следующий раз. А о чем именно – зависит от ваших пожеланий.
Ссылки
Что может биоинформатика
М. С. Гельфанд,
доктор биологических наук, кандидат физико-математических наук,
Институт проблем передачи информации РАН
«Химия и жизнь» №9, 2009

Все знают, что биоинформатика — это как-то связано с компьютерами, ДНК и белками и что это передний край науки. Более подробными сведениями может похвастаться далеко не каждый даже среди биологов. О некоторых задачах, которые решает современная биоинформатика, рассказал «Химии и жизни» Михаил Сергеевич Гельфанд (интервью записала Елена Клещенко).
Информация в биологии
В последние десятилетия появилось много новых научных дисциплин с модными названиями: биоинформатика, геномика, протеомика, системная биология и другие. Но по сути, биоинформатика, также как и, скажем, протеомика, — не наука, а несколько удобных технологий и набор конкретных задач, которые решают с их помощью. Можно говорить, что каждый человек, который определяет концентрации белков методом масс-спектрометрии или изучает белок-белковые взаимодействия, работает в области протеомики. Но не исключено, что со временем это деление станет не таким важным: применяемая технология будет менее существенной, чем способ думать, ставить вопросы. И в этом смысле биоинформатика как самая древняя из этих наук — ей целых 25 лет — играет роль цементирующего начала, потому что независимо от того, каким способом получены данные, все равно они потом попадают в компьютер. Иначе быть не может: размер бактериального генома — миллионы нуклеотидов, высшего животного — сотни миллионов или миллиарды. Транскриптомика, изучающая активность генов, получает данные о концентрациях десятков тысяч матричных РНК, протеомика — о сотнях тысяч пептидов и белок-белковых взаимодействиях. С таким количеством информации нельзя работать вручную. Мы еще помним, как печатали на бумаге нуклеотидные последовательности, потом вырезали напечатанные строчки, подставляли друг под друга и таким кустарным способом делали выравнивание — искали сходные участки. Это было возможно, когда речь шла о десятках-сотнях нуклеотидов или аминокислот, но при современном объеме данных нужны специальные инструменты. Набор таких инструментов и предоставляет биоинформатика — в практическом плане это прикладная наука, обслуживающая интересы биологов.
Поскольку моя собственная работа связана в основном с анализом геномных данных, далее речь пойдет главным образом о геномике. Объемы данных еще до появления последнего поколения секвенаторов начали обгонять закон Мура: нуклеотидные последовательности геномов накапливались быстрее, чем росла мощность компьютеров. Не будет большим преувеличением сказать, что за последние годы биология начала превращаться в науку, «богатую данными». Условно говоря, в «классической» молекулярной биологии в одном эксперименте устанавливался один биологический факт: аминокислотная последовательность белка, его функция, то, как регулируется соответствующий ген. А теперь такого рода факты получаются индустриально. Молекулярная биология движется по пути, по которому уже прошли астрофизика и физика высоких энергий. Когда имеется постоянно работающий радиотелескоп или ускоритель, проблема добычи данных решена, и на первый план выступают проблемы их хранения и обработки.
С биологией происходит то же самое, причем очень быстро, и не всегда бывает легко перестроиться. Однако те, кому это удается, оказываются в выигрыше. На нашем семинаре один биолог рассказывал, как они с коллегами изучали некий белок традиционными методами экспериментальной биологии. Это сложная задача: зная, что в клетке выполняется определенная функция, найти белок, который за нее отвечает. Они нашли этот белок, занялись его изучением и убедились, что должен существовать другой белок с подобными свойствами, поскольку наличие первого объясняет не все наблюдаемые факты. Искать второй белок на фоне первого было еще более сложно, но они справились и с этим. А затем был опубликован геном человека — и, получив доступ к его последовательности, они нашли еще дюжину таких белков...
Из этого примера вовсе не следует, что практическая молекулярная биология себя исчерпала. Скорее она научилась пользоваться новыми инструментами: интерпретировать не только полоски в геле после электрофореза, концентрации мРНК и белков или, скажем, скорость роста бактерий, но и колоссальные массивы данных, хранящиеся в компьютере. Заметим, что элемент интерпретации неизбежно присутствует и в классической биологии. Когда исследователь утверждает, что белок А запускает транскрипцию гена В, он не наблюдает напрямую, как белок взаимодействует с регуляторной областью гена, а делает такой вывод из расположения полосок на геле и других экспериментальных данных. В биоинформатике, по сути, та же ситуация, только возведенная в абсолют: готовые данные лежат в компьютере, и среди них нужно отыскать пазлы, из которых получится собрать картинку.
К области технической биоинформатики относится первичная обработка данных. Секвенатор не сам «читает» молекулы ДНК, а дает на выходе кривые флуоресценции, пики на которых еще нужно превратить в нуклеотидную последовательность. Эта задача решается каждый раз по-новому для нового устройства секвенирования, и решает ее биоинформатика. Кроме того, как уже говорилось, полученные данные надо где-то хранить, обеспечивать к ним удобный доступ и т. д. Все это чисто технические проблемы, но они очень важны.
Более сложное и интересное занятие биоинформатиков — получать на основе данных о геноме конкретные утверждения: белок А обладает такой-то функцией, ген В включается в таких-то условиях, гены С, D и Е экспрессируются в одно и то же время, а продукты их образуют комплекс. Именно этим занимаемся мы, и в этом состоит практическое применение нашей науки. Для нас пользователи — другие биологи, которым мы сообщаем интересные для них факты.
Расположение и регуляция
Как можно из последовательности нуклеотидов делать выводы о функции белков и генов? Первое соображение кажется банальным: если белок похож на какой-нибудь другой, уже изученный, то с большой вероятностью он делает примерно то же самое. На самом деле оно не так уж банально: первым серьезным успехом в этом направлении биоинформатики было утверждение, что вирусные онкогены — это «испорченные» гены самого организма.
Выполнить подобное сравнение сейчас уже несложно. Существуют банки данных по нуклеотидным и белковым последовательностям (подробнее о них рассказывалось в «Химии и жизни», 2001, №2). Общее представление о том, как это должно быть устроено, появилось в конце 80-х годов, и в этом смысле биоинформатика была готова к потоку геномных данных. Сегодня это стандартный интернет-сервис: вы загружаете свою последовательность в окошко, нажимаете кнопку, и через несколько секунд вам сообщают, на какие последовательности из этой базы она похожа.
Дальше начинаются более тонкие соображения. Известно, например, что у бактерий гены часто бывают организованы в опероны, то есть транскрибируются в виде одной матричной РНК. Есть разные эволюционные теории, которые объясняют, почему так получилось, что функционально связанные гены образуют оперон. Первая теория состоит в том, что это удобно и полезно, потому и поддерживается эволюцией. Если белки имеют общую функцию, например, отвечают за разные этапы переработки одного вещества, логично, чтобы они появлялись в клетке одновременно, по одному и тому же сигналу (естественно, что при общей мРНК и регуляция одна на всех) и в равном количестве. Второе утверждение менее тривиально и более красиво. Генам, продукты которых имеют связанные функции, выгодно находиться рядом из-за горизонтального переноса. Это очень существенный механизм эволюции бактерий: участки генома одной бактерии попадают в другую, которая благодаря этому может приобрести новые полезные признаки. Понятно, что, если в новый геном переместится лишь один ген метаболического пути, то соответствующий белок будет бесполезен: субстрата для катализируемой им реакции нет, а ее продукт, в свою очередь, некому перерабатывать. Дополнительным подтверждением этой теории служит то, что у бактерий бывают геномные локусы, в которых гены из одного метаболического пути лежат на разных цепях ДНК и потому транскрибируются в разных направлениях. Здесь точно играет главную роль повышенная вероятность совместного переноса.
Тот факт, что два гена находятся рядом в каком-то одном геноме, не очень много говорит про их функциональную связь, это может быть и случайность. Однако мы умеем отождествлять гены в разных организмах. Последовательности у них, конечно, не совпадают до нуклеотида, а могут различаться довольно значительно. Но есть некие правила, которые позволяют утверждать, что это один и тот же ген, скажем, у кишечной и у сенной палочки. Итак, если пара генов находится рядом не в одном геноме, а в пятидесяти, причем у представителей разных таксономических групп (то есть это расположение не просто унаследовано от общего предка), — это означает, что они действительно тяготеют друг к другу. Если бы эволюция не поддерживала их близкого расположения, оно не сохранилось бы. И значит, можно предположить, что они функционально связаны.
Второе соображение похоже на первое. Не все бактерии имеют одинаковый набор генов: к примеру, если ген кодирует фермент, нужный для переработки какого-то углевода, то его не будет у бактерии, которая этим углеводом не питается. Зато у бактерии, которая питается именно этим углеводом, будет весь необходимый набор: и ферменты, и белок-транспортер, переносящий углевод внутрь клетки. Функционально связанные гены присутствуют в геноме по принципу «все или ничего»: как уже говорилось, бессмысленно иметь лишь фрагмент метаболического пути, а бактерии — существа экономные, то, что не приносит пользы, из их генома быстро исчезает. Поэтому если сделать таблицу, где по строкам расположить различные гены, а по столбцам — разные геномы, и отметить плюсами и минусами гены, присутствующие или отсутствующие в данном геноме, мы увидим группы генов, обслуживающих одну и ту же функцию. И неизвестный ген с тем же набором плюсов и минусов, что у некой группы, скорее всего, можно приписать к ней же.
Третье соображение связано с регуляцией активности генов. Рядом с геном обычно присутствуют участки, с которыми взаимодействуют определенные белки — они могут запускать транскрипцию, блокировать ее, управлять ее интенсивностью, иначе говоря, от них зависит активность гена в каждый момент времени. Некоторые регуляторные участки очень хорошо опознаются по характерным последовательностям «букв», но это бывает редко. Например, участки связывания факторов транскрипции мы распознаем в геномах с невысокой точностью и вместе с правильными сайтами нагребаем кучу «мусора» — похожие коротенькие участки, которые на самом деле не имеют отношения к регуляции генов. Но поскольку совместно регулируются те гены, которые совместно работают, настоящие сайты связывания находятся перед одними и теми же генами в десятке геномов, а случайные — раскиданы там и сям, и никакой закономерности в их расположении не прослеживается. Получается мощный фильтр, позволяющий отсеять «мусор». И если перед геном с неизвестной функцией устойчиво обнаруживается знакомый сайт, будет ясно, что этот ген регулируется в составе функциональной подсистемы, которая регулируется тем же регулятором и обеспечивает ту же функцию.
Мне интереснее всего изучать эволюцию регуляторных систем, но побочным продуктом при этом бывает множество функциональных предсказаний. Исследование развивается как детектив: каждое соображение по отдельности очень мелкое, но если «улик» много и они все попадают в одну точку, то можно делать уверенные утверждения. Был случай, когда мы подробно описали регуляторную систему — фактор транскрипции, сайты его связывания, то, что это будет репрессор, а не активатор, то, что связывание будет требовать кооперативного взаимодействия двух димеров, — просто глядя на буковки генома. Впоследствии все это вплоть до деталей оказалось правильным.
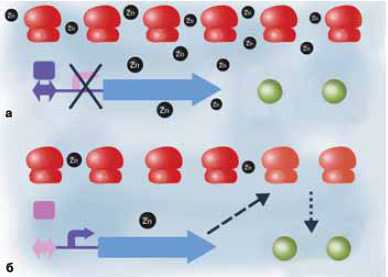
Рибосома как депо цинка
В одной из таких работ центральную роль сыграла Екатерина Панина, на тот момент студентка мехмата МГУ (потом она поступила в аспирантуру Калифорнийского университета Лос-Анджелеса и стала настоящим биологом-экспериментатором). Она пришла к нам на третьем курсе и сказала, что хочет заниматься такой биологией. К окончанию мехмата у нее было опубликовано несколько статьей в серьезных журналах.
Бактериальной клетке нужны ионы цинка: они, например, входят в состав некоторых ферментов как кофакторы. Соответственно есть и молекулярная машинерия, которая обслуживает все процессы, связанные с цинком. Мы изучали цинковый репрессор (в больших количествах цинк ядовит для клетки, поэтому выключать его транспорт при достаточных концентрациях не менее важно, чем уметь добывать его из окружающей среды), используя идеологию, о которой рассказывалось в предыдущей главке. Если перед геном имеется потенциальный сайт цинкового репрессора, то этот ген, возможно, относится к метаболизму цинка. Именно таким образом мы в свое время «вычислили» цинковый транспортер — трансмембранный белок, который обеспечивает проникновение цинка внутрь клетки.
Так вот, в 2002 году Катя обратила внимание, что потенциальные сайты цинкового репрессора почему-то часто попадаются перед генами рибосомных белков. Она поделилась этим наблюдением с научным руководителем, и я сказал, что, поскольку в геноме больше сотни генов рибосомных белков, а сайты встречались перед разными генами, это случайность. Но Катя в случайность не поверила и нашла статью Евгения Кунина (о его модели происхождения клетки см. в статье М. А. Шкроб в августовском номере), которая была опубликована незадолго до этого. Там было показано, что некоторые рибосомные белки содержат мотив связывания цинка — так называемую цинковую ленту, три или четыре цистеина на правильном расстоянии друг относительно друга и в правильном контексте. Важное наблюдение Кунина с коллегами состояло в том, что один и тот же белок в некоторых организмах имеет эти цинковые мотивы, в других — не имеет, но, судя по всему, нормально функционирует и без цинка. А у некоторых бактерий один и тот же белок имеется в двух вариантах, с цинковой лентой и без нее.
И вот Катя заметила, что в последнем случае, когда есть два варианта белка в одном геноме, тот, который без цинковой ленты, репрессируется цинковым репрессором. Иначе говоря, в присутствии цинка экспрессируется вариант белка, которому цинк нужен, а в отсутствие цинка — тот, которому он не нужен.
Основа существования любой клетки — тяжелая промышленность, производство средств производства, точно так, как нас учили на лекциях по политэкономии социализма. Около 70% белка клетки — это белки рибосом, то есть органелл, которые нужны, чтобы делать другие белки. С другой стороны, цинк — кофактор ферментов, жизненно важных для клетки, таких, например, как ДНК-полимераза. Если цинка становится мало, его полностью забирают себе рибосомные белки, ферментам ничего не остается, и клетка погибает. Но у клетки есть резервная копия рибосомного белка, которому цинк не нужен. Мы предположили, что клетка включает синтез таких белков в условиях дефицита цинка и они встраиваются в часть рибосом на место цинксодержащих белков. При этом какое-то количество цинка высвобождается. Может быть, рибосомы после этого работают чуть менее эффективно, может быть, и вообще не работают — но ради того, чтобы цинка хватило жизненно важным ферментам, которые представлены существенно меньшим числом копий, стоит пожертвовать небольшой долей рибосом.
Мы написали статью, но в течение года ни один уважаемый журнал не принял к публикации безумную теорию о рибосомах как депо цинка. Однако мне Катина находка казалась очень красивой, и я единственный раз в жизни воспользовался тем, что мой дед, как член Академии наук США, имеет право представлять статьи для публикации в «Proceedings of the National Academy of Sciences of the USA». Он послал статью на рецензию Кунину, который дал положительный отзыв (и, кажется, кому-то еще). Статья вышла в PNAS, и, как вскоре выяснилось, очень вовремя: через полгода появилась статья японских биологов, которые экспериментально показали то же самое. Можно догадаться, что они над этим работали давно, и, вероятно, им было немного обидно, что компьютерное предсказание предвосхитило их результаты.
Заметим, что вся эта история построена на очень мелких частных наблюдениях (есть в белке цистеины — нет цистеинов, есть потенциальный сайт репрессора — нет сайта...). Но в совокупности эти мелочи позволили сделать нетривиальное заключение, оказавшееся абсолютно верным. Вообще, когда мы публикуем статьи, то стараемся как можно более четко сказать, какое из наших предсказаний считаем надежным, а какое может оказаться неправильным. Так вот, среди тех, в которых мы были уверены, неправильных пока не оказалось ни одного (проверены уже десятки), а вот среди слабых проколы действительно были, хотя тоже не часто.
Отвертка со съемным жалом
Не менее красивыми были работы с белками-транспортерами (я в них участвовал только на ранних стадиях, поэтому имею полное право их хвалить, не становясь хвастуном). Транспортеры — золотое дно для биоинформатиков, поскольку опознать транспортер, в особенности бактериальный, достаточно легко. У них есть несколько гидрофобных спиралей, проходящих через мембрану: между ними находится канал, сквозь который ион или молекула, нужные для жизни клетки, проникают внутрь. Трансмембранные сегменты можно найти в белковой последовательности с помощью специальных программ. И если в неизвестном бактериальном белке пять или шесть таких сегментов, это почти наверняка транспортер (потому что другие трансмембранные белки, например участники дыхательной цепи или родопсин, хорошо известны). Остается установить, что за вещество он переносит.
Изучать специфичность транспортеров в эксперименте — удовольствие ниже среднего. С ферментами гораздо проще, это практически рутинная задача, которую можно доверить роботу. Вы гиперэкспрессируете фермент (то есть заставляете синтезироваться в больших количествах), а потом предлагаете ему пять сотен разных субстратов и смотрите, с каким из них пойдет реакция.
Транспортер, конечно, тоже можно гиперэкспрессировать. Но чтобы он заработал, он должен сразу встроиться в мембрану, иначе гидрофобные сегменты «налипнут» друг на друга, белок образует нефункциональные агрегаты. Поэтому приходится делать множество мембранных пузырьков-везикул, встраивать в них белки в правильной ориентации, а потом смотреть, попадает ли искомое вещество внутрь везикул. Вдобавок транспортеры бывают разные. Одни закачивают в клетку полезные вещества против градиента концентрации и затрачивают на это энергию молекулы АТФ, которую расщепляет специальный белок — АТФаза. Другие осуществляют вторичный транспорт — впуская «нужную» молекулу, одновременно выпускают по градиенту концентрации ион водорода, калия или натрия. Если транспортер АТФ-зависимый, то, чтобы он заработал, необходимо собирать конструкцию из нескольких белков, в том числе АТФазы. А если это вторичный транспорт, то нужно еще угадать, концентрацию какого иона надо увеличить внутри шарика. Отсюда ясно, что биохимия транспортеров — наука для сильных духом и экспериментальных данных по ним мало.
С другой стороны, определять специфичность транспортеров биоинформационными методами существенно проще. Достаточно прибегнуть к уже знакомой логике: например, если синтез этого белка регулируется цинковым репрессором, это, скорее всего, и будет цинковый транспортер, а если его ген находится в одном локусе с генами катаболизма рибозы, он, очевидно, переносит внутрь клетки рибозу... Именно таким образом мы в свое время нашли рибофлавиновый транспортер: имеется белок с неизвестной функцией, у него шесть потенциальных трансмембранных сегментов, регулируется совместно с генами рибофлавинового пути — значит, это транспортер либо рибофлавина, либо его предшественника. Но поскольку в некоторых геномах присутствовали и этот транспортер, и рибофлавин-зависимые белки, но не было пути синтеза рибофлавина из предшественников, значит, это мог быть транспортер только рибофлавина.
Проверять экспериментально конкретное предсказание существенно проще, чем начинать с нуля. Я всегда объясняю студентам, что биоинформатик — существо совершенно беззащитное, наподобие того персонажа приключенческого романа, который знает, где лежит клад. Пока он молчит, все его берегут и за ним ухаживают, но, когда он проговорится, он уже не нужен. Как только биоинформатик сказал «этот белок обладает такой-то функцией» — исключительно от порядочности экспериментаторов зависит, возьмут ли они его в соавторы после того, как проверят это утверждение. А утверждения, как читатель уже убедился, предельно простые и конкретные, достаточно один раз произнести их вслух.
С таких же простых умозаключений начиналась история более сложная, но и более интересная. Мы изучали регуляцию пути биосинтеза биотина (биотин — витамин Н, или В7, кофактор многих важных ферментов). Биотиновый транспортер был в это время не известен. У нас по ходу работы обнаружился транспортный белок, который регулируется, а иногда и локализуется вместе с генами биотинового пути. Дальше все как с рибофлавином: нашлись организмы, где биотинового пути нет, но есть белки, которые зависят него как от кофактора, и есть тот самый потенциальный транспортер — следовательно, это транспортер биотина.
Как уже было сказано, транспортеры бывают АТФ-зависимые и осуществляющие вторичный транспорт. Биотиновый транспортер был одиноким, никакого гена АТФазы поблизости не просматривалось, а значит, это был вторичный транспортер. Но затем мы увидели, что в некоторых геномах рядом с биотиновым транспортером попадаются какие-то АТФазы. Что это означает, на том этапе было непонятно, и потому мы просто упомянули про это в статье одной фразой.
Примерно тогда же мы изучали регуляцию кобаламинового пути. Кобаламин, или витамин В12, —также кофактор важных ферментов, очень крупная молекула с метаболическим путем соответственной сложности. Для этой истории существенно, что в центре молекулы кобаламина есть ион кобальта, который приносят в клетку опять же транспортеры. Таких транспортеров мы нашли немало, опубликовали о них статью — и в скором времени получили письмо от Томаса Эйтингера из Института микробиологии Гумбольдтовского университета (Берлин). Он призывал нас обратить внимание на то, что любой кобальтовый транспортер также может транспортировать никель, и наоборот, потому что специфичность у них слабая. Мы ответили, что рассматриваем транспортеры с точки зрения их функциональной роли в клетке, и если ген белка находится в одном опероне с большим набором генов кобаламинового синтеза — безусловно, белок нужен клетке как транспортер кобальта, хотя in vitro его и можно заставить переносить никель. А если мы видим ген транспортера в одном опероне с никель-зависимой уреазой, то это, безусловно, никелевый транспортер.
Намечались перспективы совместной работы, и Дмитрий Родионов, который делал эту работу, подал вместе с немецкими коллегами заявку на небольшой совместный грант и поехал на три месяца в Берлин. (Дмитрий закончил МИФИ, после чего занимался у нас геномикой; потом работал в США, а сейчас выиграл грант академической программы «Молекулярная и клеточная биология» на создание новой группы и возвращается в Москву.)
К этому времени мы с ними начали делать (по электронной почте) большой проект по сравнительной геномике транспортеров никеля и кобальта, где классифицировали их, во-первых, по регуляции, а во-вторых, по локализации, совместной с кобальтовыми или никелевыми функциональными белками. Так вот, в одном из этих никелево-кобальтовых семейств наблюдались некоторые странности. С одной стороны, АТФазы и трансмембранные белки, образующие канал для иона, как положено, располагались рядом и регулировались совместно. С другой стороны, в том же опероне мог находиться еще один трансмембранный белок. Причем эти «посторонние» белки в кобальтовых и никелевых транспортных системах отличались довольно сильно, не были гомологичными в отличие от АТФаз и трансмембранников. И вдобавок АТФаза и трансмембранный белок оказались гомологичными тем самым «лишним» биотиновым белкам, которые то попадались, то нет в предыдущем исследовании.
До сих пор не знаю, каким способом Дима уговорил немецких коллег на следующий безумный эксперимент. «Классическим» биохимикам, которые всю жизнь изучают транспорт кобальта и никеля у бактерий, он предложил: давайте у транспортера отключим АТФазу и трансмембранный белок, гомологичные биотиновым, оставим один только уникальный компонент. Ведь биотиновому транспортеру АТФаза и «основной» трансмембранник не очень нужны, они то есть, то их нет, — может быть, они и никелевому транспортеру не нужны, одинокий негомологичный трансмембранник и сам справится? Неизвестно, почему добропорядочные немецкие биохимики решились на это странное деяние: лишить вроде бы обычный АТФ-зависимый транспортер АТФазы и посмотреть, что будет. Так или иначе, Дима оказался прав. Одинокий трансмембранник работал как кобальтовый транспортер — менее эффективно, но работал. Это был первый пример двойной системы, которая, если есть АТФаза, работает какАТФ-зависимая, а если ее нет, работает как ион-зависимая.
Позднее берлинские коллеги то же самое сделали с биотином: взяли бактерию, у которой биотиновый транспортер имеет АТФазу и трансмембранник, отключили их гены — и показали, что этот белок в одиночестве тоже работает как биотиновый транспортер, хотя и с меньшей мощностью, чем в присутствии АТФазы.
Дмитрий Родионов в это время уже работал постдоком в лаборатории Андрея Остермана в Институте медицинских исследований Бэрнема в Ла-Хойе. Остерман — замечательный человек, биохимик, который понял эффективность биоинформатических методов, научился ими пользоваться и нашел с их помощью множество новых ферментов. И вот, когда Дмитрий попал в круг биохимиков и начал с ними общаться, оказалось, что подобных транспортеров, переносящих разные субстраты — кофакторы, аминокислоты, ионы, — существует несколько десятков. (Кстати, таким же оказался и рибофлавиновый транспортер.) Разные исследовательские группы независимо друг от друга изучали эти транспортеры, не имея представления о том, что они принадлежат к одному семейству.
Стало понятным и то, как возможна подобная организация. Кобальтовые и никелевые транспортеры отдельно от своей АТФазы не встречаются (если ее не убрать экспериментально). Но есть и другой класс бактериальных транспортеров, которые используют одну и ту же АТФазу — как отвертку со съемным жалом. Универсальные АТФаза и трансмембранный белок в этом случае могут кодироваться вместе с рибосомными белками, то есть экспрессируются постоянно и в больших количествах. А те белки, которые обеспечивают транспортерам специфичность, раскиданы там и сям в соответствующих оперонах. А в отсутствие АТФазы такой белок худо-бедно работает как вторичный транспортер, и поэтому в геномах некоторых организмов мы видим только его.
Биоинформатика и теория эволюции
Однако эти «прикладные» открытия — дело очень важное и полезное, но для нас, биоинформатиков, не главное. А главное, что принесла нам индустриальная революция в биологии, — появилась возможность на другом уровне обсуждать эволюцию. Даже банальные утверждения, скажем, о процентном сходстве геномов человека и шимпанзе нетактривиальны, как могут показаться. Молекулярная эволюция поучительна тем, что на ней замечательно выполняются дарвинистские представления о природе вещей.
Данные, полученные молекулярными биологами, теперь оказывают серьезное влияние на таксономию — классификацию растений и животных. Поначалу ботаники и зоологи скептически относились к молекулярным генеалогическим деревьям, показывающим степень родства между видами на основе сравнения нуклеотидных последовательностей, но надо признать, что и первые молекулярные деревья были не слишком удачными. Сейчас прямо на глазах происходит конвергенция — классическая и молекулярная таксономии движутся навстречу друг другу. Уже понятно, что молекулярные деревья, если они построены с соблюдением определенных правил, достаточно близки к реальности и вполне могут стать поводом для пересмотра ортодоксальных таксономических представлений, основанных на морфологии — сравнении внешних черт организмов. И, как ни странно, оказывается, что у видов, которых будто бы насильно помещают вместе исходя из сходства их генов, действительно отыскиваются общие признаки. Получается, что хорошее молекулярное дерево не противоречит морфологической конструкции, просто другие признаки оказываются ведущими.
Что касается бактерий, то в эпоху классической биологии их классифицировали по форме клеток и по метаболическим свойствам: какие сахара они могут утилизировать, какие аминокислоты и кофакторы могут синтезировать сами, а в каких нуждаются как в составной части внешней среды и т. п. Эта таксономия была очень слабой, поскольку у бактерий по сравнению с высшими организмами очень мало морфологических и функциональных признаков. Сегодня таксономия бактерий, по-видимому, полностью основывается на молекулярных данных. В массовом порядке пересматриваются видовые названия. Но самым впечатляющим достижением в этой области была, конечно, работа Карла Вёзе, который в 1977 году на основании молекулярной таксономии постулировал существование архебактерий (сейчас их называют археями) — третьего домена жизни, отличного от эукариот и «настоящих» бактерий.
Нельзя сказать, что все проблемы систематики бактерий отныне решены. В значительной мере оказалось разрушенным представление о том, что такое бактериальный вид. Обнаружилось, например, что у двух штаммов кишечной палочки — представителей одного вида — до трети генов могут быть уникальными, то есть присутствовать в одном штамме и отсутствовать в другом. Много неожиданного и интересного уже известно о бактериальной эволюции. В частности, оказалось, что горизонтальный перенос — обмен генетическим материалом — может происходить между таксономически далекими существами. Например, Metanosarcina — типичная архея, но треть ее генов имеют бактериальное происхождение, и эти гены обслуживают практически весь ее метаболизм, в то время как механизмы транскрипции, трансляции, репликация, устройство мембраны у метаносарцины характерны для архей. По этому примеру можно судить о том, насколько увлекательно сейчас заниматься эволюцией бактерий.
На мой взгляд, самое интересное — это эволюция регуляторных систем. Мы достаточно много знаем про эти системы у бактерий и можем представить, как меняются регуляторные системы, как локальный регулятор вдруг начинает управлять десятками генов или меняет специфичность, как перестраиваются регуляторные каскады. И это может быть очень важно с фундаментальной точки зрения, потому что здесь можно пойти гораздо дальше. Отличие человека от шимпанзе или даже от мыши едва ли обусловлено набором генов: они у млекопитающих практически одни и те же, если сравнивать по набору функций. Причина скорее в регуляции: какие гены, когда и в каких тканях активны.
Скорее всего, «скачки» эволюции, любые резкие изменения морфологических признаков обеспечиваются как раз на уровне регуляции. Мы уже знаем такие примеры у бактерий, дрожжей и других относительно простых организмов. У большинства бактерий имеется один железный репрессор, который реагирует на присутствие ионов железа и регулирует множество генов: белки, обеспечивающие запасание и транспорт железа, железозависимые ферменты. А у других бактерий есть три разных репрессора, которые эти функциональные группы поделили: одни регулируют запасание железа, другие транспорт и синтез, третьи — ферменты. Это на самом деле радикальное изменение, был один ответ на железо, а получилось три разных.
Есть замечательные экспериментальные работы, выполненные на многоклеточных. Почему морской еж единственный среди иглокожих имеет твердый скелет? Ответ предложил Эрик Дэвидсон из Калифорнийского технологического института. Он изучил регуляторный каскад, который отвечает за развитие этого скелета, а потом нашел этот каскад у морской звезды, только у нее он включается существенно позднее, поэтому развиваются лишь основания иголочек, не соединенные между собой. У ежа тот же каскад включается на какое-то количество клеточных делений раньше, соответственно захватывает большее число клеток, и развивается сплошной скелет. Таким образом, чисто регуляторное изменение дает абсолютно новый признак.
У меня есть надежда, что сравнительный анализ регуляции даст ответы на вопрос, который беспокоит палеонтологов и морфологов на нынешнем этапе развития синтетической теории эволюции: каким образом накопление мелких изменений дает радикально новые признаки? Похоже, что это можно объяснить перенастройкой регуляции. Мы уже умеем это делать на простых организмах, но рано или поздно очередь дойдет и до более сложных. И когда это случится, произойдет третий большой прорыв в этом направлении, если первым считать дарвиновский естественный отбор, а вторым — соединение эволюционной биологии с генетикой.
Я б в биоинформатики пошёл, пусть меня научат!
Биология не раз переживала новое рождение: быв сначала «полевой» наукой, изучавшей животных и растения, в XX веке она значительно переместилась в лаборатории, концентрируясь на молекулярных основах жизни и наследственности. В XXI веке история двинулась дальше: многие эксперименты теперь проводятся на компьютере, а материалом для изучения являются последовательности белков и ДНК, а также информация о строении биологических молекул. В этой статье мы дадим несколько советов тем, кто решил связать свою карьеру с компьютерной биологией, став, тем самым, биоинформатиком.
Обратите внимание!
Спонсор публикации этой статьи — Лев Макаров.
В наше время в мире никого не удивишь уже названием профессии «компьютерный биолог» или «биоинформатик», хотя еще несколько десятков лет назад эти сферы деятельности — биология и компьютеры — казались совсем непересекающимися, а еще за несколько десятков лет до того никаких компьютеров не было вовсе. Причем сейчас этот термин включает в себя уже достаточно много отдельных занятий, требующих разной подготовки и разного взгляда на науку и ее место в жизни: биоинформатик, специалист по обработке информации, разработчик баз данных, программист, куратор онтологий, специалист по молекулярному моделированию — все они занимаются разными вещами, хотя со стороны их отличить будет непросто. Все это без намеков говорит нам, что компьютеры прочно вошли в будни биологов, причем это не только е-мейл и фейсбучек, но и масса более специальных навыков, без которых исследователю сейчас и в будущем уже не обойтись (см. врезку). Студент вы или профессор, — никогда не поздно начать совершенствовать свои навыки биоинформатика !
«Сухая» биология
«Биомолекула» довольно много внимания уделяет компьютерной, или, как ее еще называют, «сухой» биологии — современной отрасли биологической науки, в которой главным инструментом исследователя является обычный компьютер. (Правда, частенько приходится прибегать к помощи и не совсем обычных — суперкомпьютеров.) На нашем сайте есть специальная рубрика, посвященная этой науке, — «“Сухая” биология» [1], — ознакомиться с которой мы и предлагаем заинтересованному читателю. В частности, там идет речь о концепции количественной биологии [2], о способах вычисления пространственной структуры [3] и динамики [4] биологических молекул (с особенным акцентом на биомембранах и мембранных белках и рецепторах [5]), а также о становлении молекулярной графики [6]. В недавних статьях было рассказано о методах изучения эволюции по молекулярным данным [7], а также о новой концепции «сухой» биологии, предсказывающей будущее биологии как науки [8].
В этой статье, основанной на переводе недавнего эссе в журнале Nature Biotechnology [10], мы приводим некоторые советы для начинающих биоинформатиков — исследователей, которые планируют заниматься изучением жизни без отрыва от клавиатуры.
Словарик компьютерных терминов
- Командная стока
- способ взаимодействия с компьютером без мышки и кнопочек, а лишь набирая в окне терминала специальные команды и оперируя информацией, хранящейся в текстовых файлах. Чаще всего командная строка ассоциируется с компьютерами под управлением UNIX/Linux, хотя и в WindowsTM, и в Mac OSTM они тоже есть.
- Кластер
- объединенные в единую высокоскоростную сеть и работающие вместе компьютеры, которые можно использовать для решения ресурсоемких задач. Обычно оснащены системой планирования задач и диспетчеризации ресурсов.
- Конвейер
- способ решения конкретных задач по обработке данных, объединяя программы более общего назначения в цепочку так, чтобы информация, выдаваемая одной программой, попадала на вход следующей.
- Исходный код (исходник)
- текст программы на одном из языков программирования. В случае интерпретируемых языков текст является программой сам по себе, а вот программу, написанную на компилируемом языке, сначала надо перевести в бинарный исполняемый файл (скомпилировать).
- Программное обеспечение (ПО)
- ну это и так понятно — добавим лишь, что это набор инструкций для компьютера, который позволяет пользователю (или программисту) решать нужные ему задачи — от набора текстов в ворде до анализа генетической последовательности или расчета молекулярной динамики.
- Скрипт
- разновидность программы, написанная на интерпретируемом языке (а значит, не требующая специальной компиляции) и используемая биоинформатиками для автоматизации своих задач, для реализации парадигмы конвейера.
- Система контроля версий
- компьютерная система управления разработкой сложных программ, включающих десятки или сотни файлов с исходниками, тысячи или даже миллионы строчек кода, и разрабатываемых несколькими или многими программистами. Позволяет программе со временем не «расползаться», а программистам — легко переключаться между разными версиями и «ветками» разработки.
- UNIX/Linux
- семейство исходно многопользовательских и многозадачных операционных систем (ОС). Чаще всего используется на серверах и вычислительных кластерах, однако может устанавливаться и на персональные компьютеры в качестве альтернативы коммерческим ОС (таким как Windows). Особенностью этих операционных систем является модель разработки — поскольку ОС имеют открытый исходный код, в их создании участвуют программисты-добровольцы со всего мира. Впрочем, число версий так велико, что есть и проприетарные («закрытые») ветви — как, например, Mac OS, которая с некоторого времени внезапно стала «потомком» UNIX-систем.
Выбор оружия за вами
Сейчас создано такое количество разнообразных биоинформатических программ, что сделать оригинальное компьютерное исследование можно, и не программируя самостоятельно; надо только выбрать подходящее ПО. Однако не стоит слишком расслабляться: чтобы получилось что-то хорошее, надо сначала как следует понять, что же эти программы делают, и какая математическая теория лежит в их основе. Вы же не пойдете в лабораторию ставить полимеразную цепную реакцию, предварительно не узнав, что это такое и для чего нужно ? Ну так вот и с компьютерами то же самое. Биоинформатические программы, по сути, являются аналогами оборудования и методик в «мокрой» молекулярно-биологической лаборатории. (Кстати, на контрасте со словом «мокрый» биоинформатические лаборатории все чаще сейчас называют «сухими» [8].) Поэтому, хотя от вас и не требуется вчитываться в каждую строчку исходного кода, представлять себе общие принципы работы программ совершенно необходимо.
Разные программы часто воплощают один и тот же теоретический подход, но все-таки адаптированы для решения разных практических задач. Например, при «сборке» генома из отдельных последовательностей ДНК [9], получаемых в результате работы автоматических секвенаторов, в случае «длинных» (сотни остатков нуклеотидов) прочтений используется алгоритм, основанный на перекрывании (Overlap-Layout-Consensus), в то время как для работы с наборами «коротких» (десятки остатков нуклеотидов) фрагментов лучше подходят графы де Брёйна. И выбор правильной программы не только сэкономит вам массу времени, но и вообще принципиально обеспечит (или не обеспечит) выполнимость поставленной задачи.
Хотя иной раз на мониторе биоинформатика и появляются занятные картинки (в данном случае — гликопротеин лихорадки Денге), чаще всего там можно увидеть текстовое окошко с непонятными колонками цифр или строчками букв.
Держите все под контролем
Одна из главных опасностей, что компьютер запросто может выдать неправильный результат, специально никак не просигнализировав об этом. Отсутствие сообщения об ошибке еще не говорит о том, что полученный результат правильный. Подав программе дикие данные на вход или просто использовав неправильные настройки, неизбежно получишь дикий ответ, и чрезвычайно важно постоянно помнить о такой возможности и уметь проверять, что полученное имеет хоть какое-то отношение к действительности. Проще всего убедиться, что все работает как следует, запустив программу для данных, ответ для которых уже известен, и убедиться, что именно он и получается. Часто для маленьких наборов данных вычисления можно провести буквально вручную, и тогда сверить ответ с получаемым на компьютере особенно занятно: если он отличается, то либо не права машина, либо вы. Но положительного результата в этом случае уже не получить — это точно.
Биохимические эксперименты никогда не проводят без отрицательных и/или положительных «контролей», так вот привыкайте и на компьютере делать то же самое. Контролем в биоинформатике последовательностей служит, как правило, проверка модели на неких случайных данных. С выбором модели генерации случайных данных надо быть очень и очень аккуратным. Дважды проверяйте, что все было без ошибок, и, главное, что полученные результаты имеют какой-то смысл, — иначе вас неизбежно подкараулят «открытия» на ровном месте.

Если программировать достаточно много, все предстает в другом свете.
Вы ученый, а не программист
Как известно, лучшее — враг хорошего. Помните, что в вашей работе важны свежие мысли и новизна результатов, а не красота исходников программы. Превосходно оформленный и документированный код, который не дает правильного ответа, несомненно, никуда не годится по сравнению с примитивным скриптом, который дает его. Другими словами, красоту в программу следует привносить только после того, как вы не раз уже убедились, что она и впрямь делает то, для чего предназначена. И — самое главное — используйте свои биологические знания по максимуму, потому что только это и делает вас компьютерным биологом. С другой стороны, полезно писать комментарии прямо по ходу написания программы: «эта функция/структура нужна для...», иначе уже через неделю вы потратите много времени, чтобы понять, что здесь происходит. Повторный запуск программы — это отличное повод для приведения кода в человеческий вид; вы просто будете делать это походу «вспоминания» вчерашней последовательности действий.
Используйте систему контроля версий
Использование контроля версий позволит более гибко управлять развитием кода, позволит легко возвращаться к предыдущим редакциям программы или переключаться между разными ветвями разработки, а также откроет возможность совместной разработки программы. Распространенные системы — такие как Git или Subversion — дадут возможность легкой публикации проекта в сети. Вы сделаете лучше прежде всего для себя, если не поленитесь написать несколько внятных README-файлов и положите их в нужные места проекта; это чрезвычайно вам поможет, если спустя месяцы или даже годы вам придется вернуться к старой программе. Документируйте программы и скрипты, чтобы было понятно, что они делают. Когда публикуете научную статью, хорошим тоном будет опубликовать также оригинальные программы, которые использовались для обсчета данных: это позволит другим использовать тот же метод и воспроизвести ваши результаты. Неплохо бы также вести электронный дневник, в котором был бы записан весь ход работы. Онлайн-репозитории, такие как Github, позволяют делать это, а также позволят вам хранить рабочие версии программы, что станет дополнительным уровнем бэкапа ваших наработок (см. таблицу 1).
| Задача | Инструменты |
|---|---|
| Совместная разработка программ | Сделайте ваш код (и, возможно, данные) доступными в сети с такими онлайн-хранилищами как Github, Sourceforge или Bitbucket. В интернете много руководств по использованию этих систем. Существуют также системы управления научными проектами, о которых рассказано в отдельной врезке. |
| Для сложных задач пишите скрипты и конвейеры | Для этого можно использовать как современные разработки, вроде Ruffus, так и проверенные временем классические UNIX-утилиты типа Make. Выбор конкретного инструментария зависит от личных предпочтений и любимого языка программирования |
| Сделайте ваши «конвейеры» доступными | Не исключено, что в командной строке вы себя чувствуете, как рыба в воде, но большинство ваших коллег, наверняка, нет. Созданные вами конвейеры можно оснащать графическими интерфейсами с помощью систем Galaxy или Taverna. |
| Инструменты разработчика (IDE) | Конечно, программы можно писать в любом текстовом редакторе, начиная с vi, но будет лучше, если вы освоите более продвинутые инструменты — такие как текстовый редактор Emacs или полнофункциональную среду разработки типа Eclipse. И, опять же, конкретный выбор будет основан на ваших предпочтениях и любимом языке программирования. |
Системы управления проектами
Еще одним полезным инструментом, помимо систем контроля версий, который можно позаимствовать из программисткой практики, являются системы управления проектами. Проще всего воспринимать их как продвинутый электронный журнал, который предоставляет вам следующие дополнительные возможности:
- Создание и назначение задач. Например, «посчитать то-то». Внутри задачи можно вести обсуждения, которые будут удобно структурированы и не превратят вашу почту в склад ужасающей переписки вроде «Re: Project X (100)» При этом, однако, можно настроить почтовые уведомления, поэтому никто не пропустит важный комментарий.
- Прикладывать и организовывать файлы с подробными описаниями и поддержкой версий а-ля Dropbox. Вам приходилось подолгу искать в нескольких ветках переписки по проекту какие-нибудь файлы с маловразумительными названиями, вроде «report_ACC_clean.xxx»?
- Во встроенную Wiki можно заносить описания процедур запуска программ, методики экспериментов, встраивать видеозаписи и даже рендерить LaTeX формулы.
- Текстовый поиск по всему содержимому, в том числе по приложенным файлам.
- Интеграция с системами контроля версий для разработки ПО позволяет удобно соотносить задачи с изменениями в репозиториях.
- Есть даже такие экзотические возможности, как организация своего аналога Google Docs для одновременного редактирования текста. Не всю информацию можно доверить сторонним ресурсам.
В нашей лаборатории мы используем Redmine — это отличная СПО-система управления проектами, под которую есть множество плагинов. Ее можно развернуть как самостоятельно, так и взять в аренду виртуальную машины с уже установленной системой. Наиболее известным проприетарным аналогом является Basecamp.
Залевский Артур, факультет биоинженерии и биоинформатики МГУ
(группа вычислительной структурной биологии).
Заразная болезнь конвейерит
Конвейер (pipeline) — программная цепочка из нескольких или многих инструкций, позволяющая проводить в точности те же операции на новом наборе данных. Конвейеры и скрипты незаменимы в работе компьютерного биолога, но они также могут загнать ваше сознание в прокрустово ложе скрипта и в корне прервать полет фантазии.
Поэтому нужно предупредить: не пишите всеобъемлющих скриптов слишком рано. Сначала убедитесь, что ваша задумка сработала, и только потом программируйте конвейер. Да и в этом случае трижды подумайте: а оно надо? Точно ли конвейер в этом случае сэкономит время и позволит вывести исследование на новый уровень? Смогут ли (и захотят ли!) этой программой пользоваться другие люди? Если в дальнейшем никто, кроме вас, этой программой пользоваться не собирается, то проще будет оставить ее на уровне работающего скрипта и не увлекаться слишком полной автоматизацией процесса. Тем более — если работа была разовая, в вряд ли заново придется делать то же самое. В этом случае достаточно просто записать в журнал проделанное и с чувством выполненного долга двигаться дальше. В любом случае, вежливо будет в статье написать «scripts are freely available at request», если вы и впрямь не против поделиться своим решением.
Полет фантазии
Ну конечно же, вы можете. Что захотите — то и можете. В том смысле, что креатив и смелая фантазия в работе компьютерного биолога совершенно необходимы, потому что иначе сделать ничего интересного не получится. Адаптируйте существующие методы, создавайте новые, предвидьте успех и не бойтесь неудачи. В этой области очень многого можно достичь, просто лазая по интернету и общаясь с коллегами в лаборатории или в сети. Самообразование не только научит вас решать конкретные проблемы — оно научит вас постоянно учиться.
Запишитесь на онлайн-курсы (см. табл. 2), но это будет только начало, а не конец обучения. Лишь смерть обрывает обучение по-настоящему творческого человека.
| Полезный навык | Ресурсы |
|---|---|
| Онлайн-курсы (Massive open online courses) | Сейчас такие курсы переживают взрыв популярности, и уже предлагают крайне широкий спектр тематик для изучения прямо через интернет. На сайтах Coursera, Udacity, edX и Kahn Academy есть масса полезного из области биоинформатики, геномики, компьютерной биологии, статистики и разнообразного моделирования. |
| Обучение программированию | Codeacademy и Code School не являются чем-то заточенным под биологию, но хорошо подходят для начал программирования. Потом можно продолжить с курсом «Python для биологов». Множество хороших примеров доступно на сайте http://software-carpentry.org. |
| Решение биоинформатических задач | Практическое изучение биоинформатики путем изучения программирования и соревнования с другими участниками проекта доступно на российском сервисе Rosalind. |
| Международные организации | GOBLET — международная организация по биоинформатическому образованию, а ELIXIR — европейское объединение, обеспечивающее различную информационную поддержку и инфраструктуру для исследований в области наук о жизни. |
| Блоги и листы подписки | В сети есть масса блогов и списков рассылки для компьютерных биологов, например http://stephenturner.us/p/edu и http://ged.msu.edu/angus/bioinformatics-courses.html. Для вычислительных химиков есть еще CCL.net. |
| «Локальные» российские ресурсы | |
| Обучение основам биоинформатики (курсы и свободное посещение) | Московская школа биоинформатики даст основные навыки в этой сфере, а курс по работе с данными высокопроизводительного секвенирования расскажет, как получают полные последовательности геномов. Институт биоинформатики в Санкт-Петербурге знакомит студентов с основами биоинформатики на примере реальных научных исследований (также проходит Летняя школа). |
| Вузы, в которых преподают биоинформатику | |
| Опыт работы с Linux/Unix | Помощь в установке и настройке одного из дистрибутивов Linux вам могут помочь в сообществах Russian Fedora или Ubuntu. Также вы можете обратиться с вопросами на http://linux.org.ru; более того, на этом ресурсе можно получить и ответы на некоторые научные вопросы. |
Никого не слушай
При отработке статистических методик часто делают такой эксперимент: генерируют большие массивы случайных данных, которые случайно же обозначают как «рабочую выборку» или «контроль». А затем к этим данным применяют статистический критерий, который должен выявить различия между данными, которые исходно не различаются, и... Для многих «выборок» p-значение частенько указывает на статистически значимое различие. Биологические наборы данных, например, полученные из геномного анализа или из скрининговых тестов, также полны случайного «шума» и часто огромны по размерам. Будьте готовы к тому, что при анализе подобных данных вам придется столкнуться с ложноположительными и ложноотрицательными результатами, а также в исходные данные может вкрасться систематическая ошибка, возникшая из-за особенностей эксперимента или экспериментатора.
Даже у биологов, искушенных в статистике, частенько возникает соблазн наплевать на осторожность и углубиться в эксперименты с программой или скриптом, давшими интересный результат. Однако тут всегда необходима осторожность, которая подсказывает, что необходимо рассматривать любой результат как потенциально ошибочный и провести дополнительные проверки на этот счет. Если один и тот же результат удается получить с помощью разных подходов, тогда уверенность в правильности каждого из них возрастет. И, тем не менее, большинство таких «открытий» требуют экспериментального подтверждения, чтобы откинуть оставшиеся сомнения.
Самое важное — что для интерпретации полученных на компьютере результатов нужно хорошее биологическое образование и чутье. И даже то, что программа или скрипт работают правильно, еще не гарантирует, что полученный результат не является артефактом или просто неверной трактовкой каких-то других явлений.

Любимые слова компьютерных биологов.
Верный инструментарий
Обязательно освойте командную строку UNIX/Linux. Бóльшая часть биоинформатических программ имеет интерфейс командной строки. На самом деле, она чрезвычайно мощная, позволяет в тонкостях контролировать рабочие задачи, запускать программы на параллельное исполнение, и, что немаловажно, контролировать работу утилит и перезапускать их прямо через текстовый терминал, хоть с мобильного телефона. Это одно из преимуществ работы биоинформатиков — работать можно где угодно, был бы под рукой компьютер или планшет, а также выход в интернет. Освойте параллельные вычисления, потому что они позволяют запускать сотни задач одновременно и многократно повышать производительность работы. Обязательно нужно уметь хоть чуть-чуть программировать, хотя выбор конкретного языка программирования не играет большой роли: у всех у них есть свои преимущества и недостатки, и иногда нужно комбинировать несколько разных языков, чтобы сделать работу быстрее.
Помните, что выбор более популярного языка позволит вам пользоваться бóльшим набором существующих библиотек и подпрограмм, которые позволят не изобретать велосипед, а сосредоточиться на своей работе. Примером такого «склада» наработок является Open Bioinformatics foundation. Старайтесь не использовать Microsoft Excel (только для вывода таблиц, которые будут читать некомпьютерные биологи, которые только с ним и умеют работать). Это хорошая программа, но для обработки большого количества данных она все-таки подходит плохо. Лучше всего хранить экспериментальные данные в структурированных текстовых файлах (хороший вариант для таблиц — csv) или в SQL-базе — это позволит получать доступ к информации прямо из вашей программы.
И, да, делайте бэкапы!
Элементарно, Ватсон!
Раз уж вы станете компьютерным биологом, вам все время придется возиться с данными. Они хранят множество историй, и выловить эти истории оттуда — ваш профессиональный долг. Однако скорее всего сделать это будет не так-то просто. Нужно постоянно держать в голове смысл проведенного эксперимента и схему анализа данных, а также денно и нощно обдумывать, какой же биологический смысл кроется в полученных результатах. И не является ли гипотетический подмеченный вами смысл тривиальным следствием ошибок анализа или артефактов в данных.
Чтобы все это имело смысл, нужно общаться с другими специалистами, которые получали эти экспериментальные данные, и стараться собрать картину по кусочкам. Предлагайте дополнительные эксперименты, которые смогут подтвердить или опровергнуть выдвинутую вами гипотезу. Станьте детективом, докопайтесь до ответа.
Кто-то это уже сделал. Так найдите их и спросите!
Какая бы хитрая не была проблема и как бы не был нов метод, всегда есть вероятность, что люди уже занимались тем, с чем пришлось столкнуться вам. Есть два сайта, на которых обсуждают возникшие в исследованиях проблемы — BioStars и SeqAnswers (а чисто программистские вопросы — Stack Overflow). Иногда можно получить дельный совет даже в твиттере. Поищите в интернете, кто в этой стране и в мире занимается похожими вопросами и свяжитесь с ними (см. таблицу 3).
| Лаборатория | Город | Чем занимаются |
|---|---|---|
| Группа молекулярного моделирования на биологическом факультете МГУ | Москва | Молекулярная динамика белков и пептидов |
| Группа вычислительной структурной биологии, биоинформатическая группа и лаборатория эволюционной геномики на факультете биоинженерии и биоинформатики МГУ | Москва |
|
| Лаборатория химической кибернетики и группа компьютерного молекулярного дизайна на химическом факультете МГУ | Москва |
|
| Лаборатория биокатализа и биотрансформаций и Отдел математических методов в биологии НИИ физико-химической биологии МГУ | Москва |
|
| Лаборатория моделирования биомолекулярных систем в Институте биоорганической химии РАН | Москва | Молекулярное моделирование биомембран и мембранных белков, а также биологически активных веществ |
| Лаборатории структурной биоинформатики и структурно-функционального конструирования лекарств в Институте биомедицинской химии РАМН | Москва | Компьютерное моделирование комплексов белков с белками и лекарствами, драг-дизайн, фармакология, изучение связей «структура—активность» |
| Учебно-Научный центр «Биоинформатика» и еще несколько биоинформатических групп в Институте Проблем Передачи Информации РАН | Москва | Системная биология, анализ пространственных структур биомолекул, сравнительная геномика.Организуют Московский биоинформатический семинар, Московскую школу биоинформатики и конференцию «Moscow Conference for Molecular Computational Biology». |
| Лаборатория системной биологии и вычислительной генетики и группа биоинформатики в Институте общей генетики РАН | Москва | Поиск функциональных мотивов (сайтов связывания транскрипционных факторов и т.д.) в последовательностях ДНК |
| Лаборатория биоинформатики и системной биологии в Институте молекулярной биологии РАН | Москва | Методы биоинформатики и поиска функциональных мотивов, предсказание предрасположенности к заболеваниям |
| Лаборатория биоинформатики в НИИ Физико-химической медицины | Москва | Проблемы метагеномики и протеомики |
| Лаборатория алгоритмической биологии Академического университета РАН | Санкт-Петербург | Проблемы «сборки» и анализа геномов |
| Лаборатория «Алгоритмы сборки геномных последовательностей» национального исследовательского университета информационных технологий, механики и оптики | Санкт-Петербург | Проблемы «сборки» и анализа геномов |
| Группа биоинформатики и функциональной геномики Института Цитологии РАН | Санкт-Петербург | Изучение функционального значения общей структуры генома |
| Лаборатории функциональной геномики и клеточного стресса и механизмов функционирования клеточного генома Института биофизики клетки РАН | Пущино |
|
| Лаборатория прикладной математики в Институте математических проблем биологии РАН | Пущино | Вторичная структура РНК, альтернативный сплайсинг |
| Лаборатория физики белка Института белка РАН | Пущино | Теоретическое и экспериментальное изучение процессов сворачивания белковых молекул |
| Отдел системной биологии Института цитологии и генетики СО РАН | Новосибирск | Постгеномная биоинформатика. Компьютерный анализ и моделирование молекулярно-генетических систем. Генные сети. Модели эволюции микроорганизмов. |
| Группа лаборатории экологической биохимии Института биологии КарНЦ РАН | Петрозаводск | Молекулярное моделирование биомембран |
| Мы отдаем себе отчет, что в одной таблице нельзя перечислить все стóящие научные группы. Если мы забыли кого-то, то с удовольствием добавим. Таблица подготовлена Еленой Чуклиной (Московский физико-технический институт / Учебно-научный центр «Биоинформатика» Института проблем передачи информации РАН). | ||
В довершение можно сказать, что в интернете есть масса форумов и юзергрупп, где можно задать интересующие вопросы. Установите себе линукс и начните изучать в онлайне что-нибудь биоинформатическое. При должном упорстве вы удивитесь, как многого можно достичь, имея просто компьютер и выход в интернет!
Статья написана по мотивам эссе в журнале Nature Biotechnology [10] при участии Артура Залевского и Елены Чуклиной.

Автор этой заметки делает вид, что моделирует на компьютере взаимодействие лиганд-связывающего домена никотинового ацетилхолинового рецептора типа α7 с одним из природных нейромодуляторов.
- In vivo — in vitro — in silico;
- Пространственно-временное моделирование в биологии;
- Торжество компьютерных методов: предсказание строения белков;
- Молекулярная динамика биомолекул. Часть I. История полувековой давности;
- Компьютерные игры в молекулярную биофизику биологических мембран;
- На заре молекулярной графики;
- Как прочитать эволюцию по генам?;
- Вычислительное будущее биологии;
- Код жизни: прочесть не значит понять;
- Nick Loman, Mick Watson. (2013). So you want to be a computational biologist?. Nat Biotechnol. 31, 996-998.
Биоинформатика - это... Что такое Биоинформатика?
Биоинформа́тика
- математические методы компьютерного анализа в сравнительной геномике (геномная биоинформатика).
- разработка алгоритмов и программ для предсказания пространственной структуры белков (структурная биоинформатика).
- исследование стратегий, соответствующих вычислительных методологий, а также общее управление информационной сложности биологических систем[1].
В биоинформатике используются методы прикладной математики, статистики и информатики. Биоинформатика используется в биохимии, биофизике, экологии и в других областях.
Основные области исследований
Анализ генетических последовательностей
Обработка гигантского количества данных, получаемых при секвенировании, является одной из важнейших задач биоинформатикиC тех пор как в 1977 году был секвенирован фаг Phi-X174, последовательности ДНК всё большего числа организмов были дешифрованы и сохранены в базах данных. Эти данные используются для определения последовательностей белков и регуляторных участков. Сравнение генов в рамках одного или разных видов может продемонстрировать сходство функций белков или отношения между видами (таким образом могут быть составлены Филогенетические деревья). С возрастанием количества данных уже давно стало невозможным вручную анализировать последовательности. В наши дни для поиска по геномам тысяч организмов, состоящих из миллиардов пар нуклеотидов используются компьютерные программы. Программы могут однозначно сопоставить (выровнять) похожие последовательности ДНК в геномах разных видов; часто такие последовательности несут сходные функции, а различия возникают в результате мелких мутаций, таких как замены отдельных нуклеотидов, вставки нуклеотидов, и их «выпадения» (делеции). Один из вариантов такого выравнивания применяется при самом процессе секвенирования. Так называемая техника «дробного секвенирования» (которая была, например, использована Институтом Генетических Исследований для секвенирования первого бактериального генома, Haemophilus influenzae) вместо полной последовательности нуклеотидов даёт последовательности коротких фрагментов ДНК (каждый длиной около 600—800 нуклеотидов). Концы фрагментов накладываются друг на друга и, совмещённые должным образом, дают полный геном. Такой метод быстро даёт результаты секвенирования, но сборка фрагментов может быть довольно сложной задачей для больших геномов. В проекте по расшифроке генома человека сборка заняла несколько месяцев компьютерного времени. Сейчас этот метод применяется для практически всех геномов, и алгоритмы сборки геномов являются одной из острейших проблем биоинформатики на сегодняшний момент.
Другим примером применения компьютерного анализа последовательностей является автоматический поиск генов и регуляторных последовательностей в геноме. Не все нуклеотиды в геноме используются для задания последовательностей белков. Например, в геномах высших организмов, большие сегменты ДНК явно не кодируют белки и их функциональная роль неизвестна. Разработка алгоритмов выявления кодирующих белки участков генома является важной задачей современной биоинформатики.
Биоинформатика помогает связать геномные и протеомные проекты, к примеру, помогая в использовании последовательности ДНК для идентификации белков.
Аннотация геномов
В контексте геномики аннотация — процесс маркировки генов и других объектов в последовательности ДНК. Первая программная система аннотации геномов была создана в 1995 году Оуэном Уайтом (англ. Owen White), работавшим в команде, секвенировавшей и проанализировавшей первый декодированный геном свободноживущего организма, бактерии Haemophilus influenzae. Доктор Уайт построил систему для нахождения генов, тРНК и других объектов ДНК и сделал первые обозначения функций этих генов. Большинство современных систем работают сходным образом, но эти программы постоянно развиваются и улучшаются.
Вычислительная эволюционная биология
Эволюционная биология исследует происхождение и появление видов, также как их развитие с течением времени. Информатика помогает эволюционным биологам в нескольких аспектах:
- изучать эволюцию большого числа организмов, измеряя изменения в их ДНК, а не только в строении или физиологии;
- сравнивать целые геномы (см. BLAST), что позволяет изучать более комплексные эволюционные события, такие как: дупликация генов, латеральный перенос генов, и предсказывать бактериальные специализирующие факторы;
- строить компьютерные модели популяций, чтобы предсказать поведение системы во времени;
- отслеживать появление публикаций, содержащих информацию о большом количестве видов.
Область в компьютерных науках, которая использует генетические алгоритмы, часто путают с компьютерной эволюционной биологией. Работа в этой области использует специализированное программное обеспечение для улучшения алгоритмов и вычислений и основывается на эволюционных принципах, таких, как репликация, диферсификация через рекомбинацию или мутации, и выживании в естественном отборе.
Оценка биологического разнообразия
Биологическое разнообразие экосистемы может быть определено как полная генетическая совокупность определённой среды, состоящая из всех обитающих видов, была бы это биоплёнка в заброшенной шахте, капля морской воды, горсть земли или вся биосфера планеты Земля. Для сбора видовых имён, описаний, ареала распространения, генетической информации используются базы данных. Специализированное программное обеспечение применяется для поиска, визуализации и анализа информации, и, что более важно, предоставления её другим людям. Компьютерные симуляторы моделируют такие вещи, как популяционная динамика, или вычисляют общее генетическое здоровье культуры в агрономии. Один из важнейших потенциалов этой области заключается в анализе последовательностей ДНК или полных геномов целых вымирающих видов, позволяя запомнить результаты генетического эксперимента природы в компьютере и возможно использовать вновь в будущем, даже если эти виды полностью вымрут.
Часто из области рассмотрения биоинформатики выпадают методы оценки других компонентов биоразнообразия — таксонов (в первую очередь видов) и экосистем. В настоящее время математические основания биоинформационных методов для таксонов представлены в рамках такого научного направления как фенетика, или численная таксономия. Методы анализа структуры экосистем рассматриваются специалистами таких направлений как системная экология, биоценометрия.
Основные биоинформационные программы
- ACT (Artemis Comparison Tool) — геномный анализ
- Arlequin — анализ популяционно-генетических данных
- BioEdit — редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- BioNumerics — коммерческий универсальный пакет программ
- BLAST — поиск родственных последовательностей в базе данных нуклеотидных и аминокислотных последовательностей
- Clustal — множественное выравнивание нуклеотидных и аминокислотных последовательностей
- DnaSP — анализ полиморфизма последовательностей ДНК
- FigTree — редактор филогенетических деревьев
- Genepop — популяционно-генетический анализ
- Genetix — популяционно-генетический анализ (программа доступна только на французском языке)
- JalView — редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- MacClade — коммерческая программа для интерктивного эволюционного анализа данных
- MEGA — молекулярно-эволюционный генетический анализ
- Mesquite — программа для сравнительной биологии на языке Java
- Muscle — множественное сравнение нуклеотидных и аминокислотных последовательностей. Более быстрая и точная по сравнению с ClustalW
- PAUP — филогенетический анализ с использованием метода парсимонии (и других методов)
- PHYLIP — пакет филогенетических программ
- Phylo_win — филогенетический анализ. Программа имеет графический интерфейс.
- PopGene — анализ генетического разнообразия популяций
- Populations — популяционно-генетический анализ
- PSI Protein Classifier — обобщение результатов, полученных с помощью программы PSI-BLAST
- Seaview — филогенетический анализ (с графическим интерфейсом)
- Sequin — депонирование последовательностей в GenBank, EMBL, DDBJ
- SPAdes — сборщик бактериальных геномов
- SplitsTree
- T-Coffee — множественное прогрессивное выравнивание нуклеотидных и аминокислотных последовательностей. Более чувствительное, чем в ClustalW/ClustalX.
- UGENE — свободный русскоязычный инструмент, множественное выравнивание нуклеотидных и аминокислотных последовательностей, филогенетический анализ, аннотирование, работа с базами данных.
- Velvet — сборщик геномов
Биоинформатика и вычислительная биология
Под биоинформатикой понимают любое использование компьютеров для обработки биологической информации. На практике, иногда это определение более узкое, под ним понимают использование компьютеров для обработки экспериментальных данных по структуре биологических макромолекул (белков и нуклеиновых кислот) с целью получения биологически значимой информации. В свете изменения шифра научных специальностей (03.00.28 "Биоинформатика" превратилась в 03.01.09 "Математическая биология, биоинформатика") поле термина "биоинформатика" расширилось и включает все реализации математических алгоритмов, связанных с биологическими объектами.
Термины биоинформатика и «вычислительная биология» часто употребляются как синонимы, хотя последний чаще указывает на разработку алгоритмов и конкретные вычислительные методы. Считается, что не всякое использование вычислительных методов в биологии является биоинформатикой, например, математическое моделирование биологических процессов — это не биоинформатика.[источник не указан 792 дня]
Биоинформатика использует методы прикладной математики, статистики и информатики. Исследования в вычислительной биологии нередко пересекаются с системной биологией. Основные усилия исследователей в этой области направлены на изучение геномов, анализ и предсказание структуры белков, анализ и предсказание взаимодействий молекул белка друг с другом и другими молекулами, а также реконструкция эволюции.
Биоинформатика и её методы используются также в биохимии, биофизике, экологии и в других областях. Основная линия в проектах биоинформатики — это использование математических средств для извлечения полезной информации из «шумных» или слишком объёмных данных о структуре ДНК и белков, полученных экспериментально.
Структурная биоинформатика
К структурной биоинформатике относится разработка алгоритмов и программ для предсказания пространственной структуры белков. Темы исследований в структурной биоинформатике:
- Рентгеноструктурный анализ (РСА) макромолекул
- Индикаторы качества модели макромолекулы, построенной по данным РСА
- Алгоритмы вычисления поверхности макромолекулы
- Алгоритмы нахождения гидрофобного ядра молекулы белка
- Алгоритмы нахождения структурных доменов белков
- Пространственное выравнивание структур белков
- Структурные классификации доменов SCOP и CATH
- Молекулярная динамика
Примечания
См. также
Лекции по биоинформатике / Habr

Рад представить вам 27 лекций по биоинформатике (включая описания, слайды и полные видеозаписи). Лекции читались на летней школе по биоинформатике, которая проходила в июле 2014 под Санкт-Петербургом, собрала вместе 100 студентов со всей России и СНГ, а также лекторов из МГУ, МФТИ, Сколтеха, РАН, СПбАУ, СПбГПУ, СПбГУ, Yale University, Fox Chase Cancer Center, George Washington University, Pennsylvania State и других прекрасных организаций.
Школу традиционно проводила команда Института биоинформатики, а поддерживали ее СПбАУ РАН, СПбГУ, JetBrains, РВК, BIOCAD, EMC, Фонд «Династия» и РФФИ, благодаря чему удалось сделать мероприятие абсолютно бесплатным для всех участников.
В этом году школа пройдёт 20-25 июля под Москвой (информация тут, дедлайн скоро), все лекции также будут записываться.
Организация материалов
Если список ниже покажется вам слишком сложным для поиска и выбора лекций, то можете воспользоваться также программкой школы, плейлистом на ютубе и страницой со всеми слайдами.
1. Введение в биоинформатику (Алла Лапидус, СПбАУ РАН, СПбГУ)
[Видео] [Слайды]
Революция в ядерной физике привела много лет назад к накоплению огромного количества данных, которые надо было хранить и обрабатывать. Это оказалось под силу только компьютерам, а за ними и супер-компьютерам.
Бум геномики последних 10-15 лет продолжил эту традицию и приумножил ее: медико-биологические исследования касаются каждого из нас, а значит и данных будет производиться все больше и больше особенно в свете идеи персонализированной медицины и требований большой фармы. Тут уж без компьютерных знаний и программных продуктов и вовсе делать нечего. Но кроме того, надо хорошо знать что изучать, как, как анализировать данные и насколько им можно верить. Как хранить и обрабатывать. Где применять и где использовать.
В лекции освещено большинство этих «как». Алла ставит своей целью рассказать о значимости и широте приложений биоинформатики.

2. Мутационный процесс и методы его изучения (Алексей Кондрашов, МГУ)
[Видео] [Слайды]
Мутационный процесс — первый из двух необходимых факторов дарвиновской эволюции. В лекции рассмотрены причины и механизмы возникновения мутаций, методы измерения параметров мутационного процесса на малых, средних и больших временах, данные о скоростях мутирования и простейшие модели влияния мутирования на генетическую структуру популяции.
3. Естественный отбор и методы его изучения (Алексей Кондрашов, МГУ)
[Видео] [Слайды]
Естественный отбор — второй из двух необходимых факторов дарвиновской эволюции. В лекции рассмотрены причины и механизмы возникновения отбора, методы и параметры, применяемые для его описания и изучения, данные об отборе в природе и простейшие модели влияния отбора на популяцию.

4. Детское развитие и биоинформатика: задачи и решения (Елена Григоренко, Yale University)
[Видео] [Слайды]
В лекции рассказано о нескольких «стыках» наук о развитии и биоинформатики.
Обсуждаются проблемы пренатальной диагностики и пренатального секвенирования, а также экзомного секвенирования новорожденных.
Рассказывается об изучении влияния ранней среды развития на состояние метилома, и о геномной этиологии детских расстройств развития. В заключение рассмотрены этические вопросы, связанные с использованием геномной информации в принятии диагностических и индивидуализированных решений о развитии ребенка.
5. Секвенирование нового поколения: принципы, возможности и перспективы (Мария Логачева, МГУ)
[Видео] [Слайды]
Секвенирование нового поколения (NGS) преобразило многие области биологических и биомедицинских исследований. Оно позволяет относительно быстро и недорого получать последовательности генов и геномов ранее не изученных видов, а также – на материале большого числа индивидуумов одного вида – выявлять внутривидовую изменчивость, проводить поиск генов, ассоциированных с интересующими признаками. Помимо собственно определения последовательностей геномов NGS позволяет проводить детальный анализ экспрессии генов в разных тканях организма или при разных условиях, широко используется в эпигенетических исследованиях.
В лекции дан обзор основных методов секвенирования, их физико-химические принципы, особенности пробоподготовки, характеристика получаемых данных, их стоимость и типичные ошибки. Особое внимание уделено применимости разных методов для решения биологических задач, и даны рекомендации по планированию экспериментов, связанных с NGS.
6. Структурная биология белка: обзор проблем и подходов (Павел Яковлев, BIOCAD)
[Видео] [Слайды]
Использование только первичных последовательностей позволяет решить большинство вопросов, связанных с нуклеиновыми кислотами (ДНК и РНК). При изучении функций белков знание только первичной последовательности уже не позволяет решить большинство задач. Какие белки будут взаимодействовать между собой и как сильно? Повлечет ли замена аминокислоты смену функции белка? Как убрать побочные эффекты от лекарственного белка или увеличить его эффективность? На эти вопросы призвана ответить область биоинформатики, занимающаяся разработкой алгоритмов для моделирования пространственной формы белков и их взаимодействий.

7. De novo сборка транскриптомов (Артем Касьянов, МФТИ)
[Видео] [Слайды]
В связи со значительным удешевлением и повышением производительности технологий число проектов, посвященных de novo секвенированию геномов немодельных организмов, значительно возросло. В ряде случаев de novo секвенирование и сборка генома затруднена — к примеру, в случае его значительных размеров. В таких случаях прибегают к изучению транскриптома. Также de novo анализ транскриптома может понадобиться в случае изучения видов с большим количеством альтернативно сплайсирующихся генов, так как даже при наличии генома достаточно сложно определить полный перечень изоформ.
Лекция посвящена вопросам сборки транскриптомных данных в отсутствии генома. Рассмотрены такие темы, как сплайс-графы, программы trinity и newbler, сравнение и анализ сборок, сборка транскриптомов полиплоидных организмов.
8. Эволюция алгоритмов сборки генома (Антон Банкевич, СПбАУ РАН)
[Видео] [Слайды]
На данный момент существует уже несколько поколений методов секвенирования ДНК. Однако новые технологии бессмысленны без алгоритмов, способных обработать их результаты. Постоянно возникающие новые методы секвенирования ставят всё новые алгоритмические задачи. Одной из важнейших таких задач является сборка генома. В лекции рассказано об эволюции методов секвенирования и алгоритмических подходах к сборке генома, возникавших и продолжающих возникать с каждым шагом этой эволюции.

9. Введение в молекулярную биологию и генетику (Павел Добрынин, СПбГУ)
[Видео] [Слайды]
Лекция посвящена структуре и организации ДНК у прокариот и эукариот, молекулярным механизмам, отвечающим за сохранение и воспроизведение генетического материала. Разобраны основные механизмы, стоящие за генетической изменчивостью, и варианты реализации генетического материала.
10. Задача множественного локального выравнивания и построения синтенных блоков (Илья Минкин, Pennsylvania State University)
[Видео] [Слайды]
В лекции рассматривается две похожие алгоритмические задачи в сравнительной геномике: множественное локальное выравнивание и построение синтенных блоков. Эти алгоритмы играют крайне важную роль в сравнении полных последовательностей геномов. Рассказано о постановке задач и о базовых идеях, на которых построены некоторые современные алгоритмы.

11. Зачем и как делать презентации (Андрей Афанасьев, iBinom)
[Видео] [Слайды]
В лекции обсуждаются типы презентаций, зачем они на самом деле нужны, и рассказывается, как выступить так, чтобы слушатели все поняли и не заснули, а также каких ошибок надо избегать и с кого брать пример при подготовке своего выступления.
12. Бизнес в биоинформатике (Андрей Афанасьев, iBinom)
[Видео] [Слайды]
В лекции рассказано, какие биоинформатические компании существуют в России и в мире, кто их создал и на чем именно они зарабатывают деньги.
Обсуждены планы крупных игроков и тренды в индустрии.
В завершающей части лекции Андрей дает пищу для размышлений об организации собственного стартапа или выборе нового места работы.
13. Перспективы и проблемы системной биологии (Илья Серебрийский, Fox Chase Cancer Center)
[Видео] [Слайды]
Лекция призвана дать общее представление о системных свойствах биологических объектов. Илья Серебрийский рассказывает об основных составляющих системной биологии, об интерактомике и построении моделей, об основных проблемах в системной биологии и попытках их разрешения. Обсуждаются некоторые достижения системной биологии (главным образом из области онкологии). Также рассматриваются общедоступные ресурсы для системной биологии (TCGA/cBioPortal, CCLE).

14. Лабораторная по системной биологии (Илья Серебрийский, Fox Chase Cancer Center)
[Видео] [Слайды]
Занятие посвящено построению сетей взаимодействия на основе общедоступных баз данных. Использованы такие базы данных и веб-сервисы, как Entrez, GeneMANIA, BioGRID и другие. Рассмотрены различные методы визуализации сетей взаимодействия, в частности с помощью программы Cytoscape.
15. Метагеномика (Алла Лапидус, СПбАУ РАН)
[Видео] [Слайды]
Микробы везде, микробы правят миром, но далеко не со всеми из них мы можем познакомиться в лабораторных условиях. Подавляющее большинство из них мы не знаем как вырастить, а значит, их надо как-то извлекать из их естественной среды обитания — земли, воды, из-под корней деревьев и т.д., где они живут большими группами.
Метагеномика и помогает в этих весьма запутанных исследованиях. А еще она помогает кормить, согревать, лечить людей и ловить преступников. Всему этому и биоинформатике в метагеномике и была посвящена эта лекция.

16. Проблема проверки множества статистических гипотез (Антон Коробейников, СПбГУ, СПбАУ РАН)
[Видео] [Слайды]
В лекции рассмотрена классическая проблема проверки множества гипотез одновременно. Подобного рода задачи встают сплошь и рядом, например, при полногеномном поиске ассоциаций или анализе данных микрочипов. Рассмотрены возможные варианты решения этой проблемы, начиная от классического подхода Бонферрони и заканчивая методами, позволяющими контролировать FDR (false discovery rate).
17. Как правильно и неправильно использовать статистику (Никита Алексеев, СПбГУ, George Washington University)
[Видео] [Слайды]
Лекция посвящена ошибкам в применении статистики и способам их предотвращения. В частности, дан ответ на вопрос: в каких ситуациях можно использовать стандартные критерии для сравнения типичных представителей выборки, и что делать, если стандартные критерии не подходят?

18. Математические модели регуляции экспрессии гена (Мария Самсонова, СПбГПУ)
[Видео] [Слайды]
Понимание тонких механизмов регуляции активности генов ‒ необходимое условие для расшифровки механизмов возникновения болезней у человека. К сожалению, на сегодняшний день такое понимание отсутствует: мы не можем удовлетворительно объяснить, ни каким образом группы транскрипционных факторов взаимодействуют друг с другом, с белками хроматина, другими адапторными белками и комплексом РНК‒полимеразы, ни как и почему тот или иной участок последовательности ДНК может контролировать сложную, ограниченную в пространстве и детерминированную во времени картину экспрессии гена.
Математическое моделирование помогает понять механизмы генной регуляции путем механистического и количественного описания этого процесса. В лекции рассмотрены два наиболее распространенных подхода к моделированию экспрессии генов ‒ основанные на нелинейных уравнениях реакции‒диффузии и термодинамическом равновесии. Последовательно рассмотрены этапы построения таких моделей и приведены примеры их использования для генерации новых знаний.

19. Полулокальное и локальное выравнивание последовательностей (Александр Тискин, University of Warwick)
[Видео] [Слайды]
Вычисление наибольшей общей подпоследовательности (longest common subsequence, LCS) двух строк — одна из классических алгоритмических задач, имеющая широкое применение как в информатике, так и в вычислительной биологии, где она известна как «глобальное выравнивание последовательностей». Во многих приложениях необходимо обобщение этой задачи, которое мы называем вычислением полулокальной LCS (semi-local LCS), или «полулокальным выравниванием». В этом случае требуется вычислить LCS между строкой и всеми подстроками другой строки, и/или между всеми префиксами одной строки и всеми суффиксами другой. Помимо важной роли этой обобщенной задачи в строковых алгоритмах у нее обнаруживаются неожиданные связи с алгеброй полугрупп и вычислительной геометрией, с сетями сравнений (comparison networks), а также практические приложения в вычислительной биологии. Кроме того, задача вычисления полулокальной LCS может использоваться в качестве гибкого и эффективного подхода к (полностью) локальному выравниванию биологических последовательностей.
В лекции представлено эффективное решение задачи вычисления полулокальной LCS и дан обзор основных сопутствующих результатов и приложений. В их числе динамическая поддержка LCS; быстрое вычисление клик в некоторых специальных графах; быстрое сравнение сжатых строк; параллельные вычисления на строках.

20. Анализ семейств молекулярных последовательностей (Сергей Нурк, СПбАУ РАН)
[Видео] [Слайды]
При решении самых разных задач, от поиска регуляторных мотивов до предсказания функций белков, биоинформатикам приходится работать с целыми «семействами» эволюционно связанных нуклеотидных или аминокислотных последовательностей. В лекции рассмотрены различные способы представления таких семейств, применяемые в популярных биоинформатических инструментах и базах данных. Рассказано, как расшифровать PROSITE pattern и проинтерпретировать sequence logo, в чем отличие profile HMM от PSSM, а также как избежать ошибок при их построении и анализе результатов.

21. Эпигеномика, РНК и все такое (Андрей Миронов, ИППИ РАН)
[Видео] [Слайды]
В лекции дан обзор понятия эпигенетики. Рассмотрены уровни структурной организации хроматина, рассказано о различных эпигеномных модификациях: модификациях гистонов, метилировании CpG-мотивов. Обсуждено их влияние на экспрессию генов.
Также рассмотрена роль эпигеномных модификаций в сплайсинге, импринтинге и т.п.
Рассказано о системе XIST (X-inactivation specific transcript), антисмысловых РНК, сплайсинге, РНК-зависимой регуляции.
Также рассмотрены модели для изучения эпигеномных модификаций.
22. Контроль качества данных NGS (Константин Оконечников, Max Planck Institute for Infection Biology)
[Видео] [Слайды]
В лекции описаны погрешности секвенирования, характерные для технологий NGS. Примерами таких ошибок являются ПЦР-амплификация, сиквенс-специфичные ошибки прочтения, неравномерное распределение GC-состава и прочие. Разобраны различные методы оценки этих погрешностей и учета их при анализе. Затронут вопрос практических методов решения и существующих программных инструментов.

23. Контроль качества данных NGS, семинар (Константин Оконечников, Max Planck Institute for Infection Biology)
[Видео] [Слайды]
В ходе семинара участники научились применять навыки программирования для контроля качества данных NGS. Были рассмотрены форматы данных BAM/SAM, библиотеки pysam и pyplot, фундаментальные понятия. В частности, разобраны примеры подсчета GC-состава, оценки частоты дупликаций, распределения длины вставки, расчета покрытия в окнах.
24. Практическое секвенирование РНК (Константин Оконечников, Max Planck Institute for Infection Biology)
[Видео] [Слайды 1] [Слайды 2]
На семинаре разбиралась практическая задача анализа данных РНК-секвенирования.
В формате презентации и практики были обсуждены и продемонстрированы методы: выравнивания ридов, первоначального контроля качества, пайплайны для изучения экспрессии генов DESeq и Cufflinks, нахождение изоформ транскриптов, поиск гибридных генов.

25. Биоинформатические подходы к изучению и лечению рака на примере рака легких (Мария Шутова, ИОГен РАН)
[Видео] [Слайды]
Рак — одно из самых распространенных и опасных заболеваний. Его называют «болезнью генома» за огромный вклад накопленных и новых мутаций в его появление и развитие. При этом известно, что не только состояние генома, но и транскрипционный и даже эпигенетический статус первичных раковых клеток, а также сложный гомеостаз растущей опухоли напрямую влияют на ее свойства и, главное, восприимчивость к терапии. Единственную возможность разобраться в этом клубке взаимозависимых факторов дает биоинформатика. В лекции разобраны основные вопросы, связанные с изучением опухолеобразования, и возможные способы ответить на них с использованием биоинформатических подходов.

26. Новые омики в биологии человека: метаболомика и липидомика (Филипп Хайтович, Сколтех)
[Видео] [Слайды]
Секвенирование человеческого генома, изучение человеческих генетических вариаций, секвенирование метагенома человека, транскриптомный анализ человеческих тканей — все эти биологические методы в приложении к «big data» дали ученым большой объем ценной информации о том, что отличает человека от других животных.
Эта лекция посвящена новым «омикам», позволяющим ответить на вопросы о человеческом организме при изучении мозга и других тканей — метаболомике и липидомике.

27. Геномная сборка: взгляд в завтрашний день (Андрей Пржибельский, СПбАУ РАН)
[Видео] [Слайды]
В последние годы технологии секвенирования нового поколения сделали заметный шаг вперед: появились IonTorrent и Pacific Biosciences, Ilumina создала ряд новых протоколов. Но, как оказывается, всего этого недостаточно для того, чтобы считать проблему сборки геномов решенной. Для того чтобы пройти путь от извлечения ДНК до полностью завершенного генома, как правило, требуются десятки различных специалистов, сотни тысяч долларов и годы работы. Поэтому сегодня эта задача остается актуальной как с точки зрения биотехнологий, так и с точки зрения биоинформатики. В лекции рассмотрены последние прорывы в методах сборки геномов, новейшие типы данных, которые, возможно, позволят вывести эту задачу на новый уровень, и перспективы геномики в ближайшем будущем.
Вместо заключения
Ещё 28 лекций с позапрошлой (самой первой) летней школы по биоинформатике можно посмотреть… почему-то на мегамозге. Там же оказался и отчётный пост от одного из участников школы. И недавно мы писали, как были организованы научные проекты.
Спасибо за внимание. Всем биоинформатики!

от статистики до генетических конструкций / Habr
Чтобы погрузиться в относительно новую для себя научную область, существует огромное количество самых разных мероприятий и проектов. В последние годы их количество и форматы значительно расширились: это открытые лекции и целые научные фестивали, онлайн-курсы и онлайн-программы, летние стажировки и школы, неформальные лекции в барах, опенсорсные проекты и так далее.Уже пять лет Институт биоинформатики собирает ученых-биоинформатиков и студентов со всей страны и в течение недельной интенсивной учебы за городом на летней школе направляет биологов, медиков, информатиков и математиков в сторону биоинформатики — до сих пор очень динамично развивающейся области. С 2013 года мы записываем лекции на видео и собираем подборку полезных материалов для тех, кто не участвует в мероприятиях, но хотел бы развиваться в этой области.
Программа школы разрабатывается таким образом, чтобы объединить мир биологии и программирования и стимулировать не только развитие профессиональное развитие, но и междисциплинарное общение.

Мы продолжаем делиться архивом видеозаписей лекций летних школ. Лекции, которые можно смотреть без дополнительной подготовки, отмечены «*». Просмотр остальных лекций требует знаний в области биологии и программирования. Под катом описание содержания лекций, ссылки на слайды и видеозаписи.
Статистика в биоинформатике

Статистический анализ биомедицинских данных (Михаил Пятницкий, НИИ биомедицинской химии им. Ореховича)
Видео | Слайды
Лекция посвящена практическим аспектам статистического анализа '-омиксных' данных. В частности, описаны методики разведочного анализа, распознавания образов, кластерного анализа.
Как работать с данными и не чувствовать беспомощность? (Никита Алексеев, George Washington University)
Видео | Слайды
С одной стороны, естественные науки предоставляют огромные объемы данных и задают самые разные вопросы относительно этих данных. С другой стороны, статистика располагает множеством методов для решения таких вопросов. Такое изобилие, естественно, привносит с собой сложности – как выбрать метод, который подходит для решения именно вашей проблемы, как учесть все нюансы и не запутаться во всем этом. Универсального рецепта нет. В лекции обсуждаются различные подходы к этой проблеме.
Как правильно задать вопрос знакомому статистику (Никита Алексеев, постдок, George Washington University)
Видео | Слайды
Лекция будет полезна всем, кто сталкивается с проблемами статистической обработкой данных. Какие для них возможны решения, какие возникают трудности, и что спрашивать у статистика, с которым удалось начать сотрудничать, чтобы получить максимальную пользу для своего проекта.
Иммуноинформатика

Анализ репертуаров иммунных рецепторов (Вадим Назаров, Высшая Школа Экономики, Институт Биоорганической Химии РАН)
Видео | Слайды
Применение NGS технологий в иммунологии позволило очень глубоко секвенировать репертуары клеточных рецепторов. Но на полученные данных, к сожалению, нельзя просто смотреть и получать инсайты – необходимо разработать различные методы анализа репертуаров. О том, какие методы были разработаны, насколько они адекватны, куда движется этот мир, и где в нем можно себя приложить.
Иммуноинформатика: алгоритмический подход к решению прикладных задач иммунологии (Яна Сафонова, Центр алгоритмической биотехнологии, СПбГУ)
Видео | Слайды
Анализ адаптивной иммунной системы является важнейшим этапом при разработке лекарств, оценке эффективности лечения, изучении различных заболеваний. Современные NGS технологии позволили делать глубокое сканирование репертуаров антител и Т-клеточных рецепторов, что способствовало развитию новой области биоинформатики: иммуноинформатика.
Иммуноинформатика решает задачи, имеющие применение в различных иммунологических направлениях: мониторинг развития иммунного ответа, анализ эволюционного развития репертуаров, понимание разнообразия адаптивной иммунной системы. В рамках лекции рассматриваются задачи современной иммуноинформатики и обсуждаются перспективы ее развития.

Молекулярное баркодирование, анализ репертуаров Т-клеточных рецепторов и антител (Дмитрий Чудаков, Заведующий лаборатории геномики адаптивного иммунитета в Институте биоорганической химии РАН, руководитель группы адаптивного иммунитета в CEITEC MU, Masaryk University)
Видео | Слайды
Высокопроизводительное секвенирование интересующих фрагментов генома (targeted resequencing) потенциально позволяет проводить глубокий анализ, выявляющий присутствие в образце редких подвариантов последовательностей, а также дающий полную картину о структуре разнообразия последовательностей в образце.
Однако, «бутылочные горлышки» на стадиях получения и приготовления образцов для массированного секвенирования, количественные искажения, связанные со стохастической природой ПЦР, неравной эффективностью амплификации и секвенирования различных последовательностей, а также накопление ошибок ПЦР и собственно секвенирования, существенно ограничивают возможности такого анализа.
Уникальное молекулярное баркодирование (unique molecular bacrodes, unique molecular identifiers, UMI) позволяет радикально повысить качество секвенирования, в том числе протяженного, эффективно корректировать накопленные ошибки без потерь реального разнообразия вариантов, устранить количественные искажения, а также практически идеально нормировать образцы для сравнительного анализа.
В лекции рассказывается о том, как работают подходы на основе молекулярного баркодирования с примерами из личного опыта работы с репертуарами рецепторов иммунных клеток – Т-клеточных рецепторов и антител.
Системная биология
Введение в системную биологию (Илья Серебрийский, Fox Chase Cancer Center, USA)
Видео | Слайды
В лекции дается общее представление представление о системных свойствах биологических объектов. Краткое описание основных составляющих системной биологии. Интерактомика, построение моделей. Некоторые достижения системной биологии (выборочно, в основном в области онкологии) и соответствующие общедоступные ресурсы (TCGA/cBioPortal, CCLE)

Вычислительная системная биология для изучения и лечения рака (Андрей Зиновьев, Institut Curie)
Видео | Слайды
Вычислительная системная биология рака является применением общих подходов системной биологии, связанных с системным сбором полногеномных данных и их математическим моделированием, для изучения канцерогенеза, прогнозирования и разработки новых методов лечения раковых заболеваний. Данных подход связан с рядом особенностей таких как учет быстрой эволюции биологической системы в условиях геномной и эпигеномной нестабильности, взаимодействия с клетками нормальной стромы и воздействия различных факторов межклеточной среды, разнообразия и качества клинического материала. В лекции кратко описаны несколько характерных подходов к анализу и моделированию данных в биологии рака. В частности, принципы формализации и использования в моделировании знания о биохимии рака (Атлас Сигнальных Сетей в Раке), подходы к деконволюции полногеномных молекулярных профилей в раке, построение дискретных математических моделей с целью предсказания эволюции раковой опухоли.
Проблема воспроизводимости результатов в системной и не только биологии (Илья Серебрийский, Fox Chase Cancer Center, USA)
Видео | Слайды
Проблема воспроизводимости результатов – ключевая для современной биологии, особенно для системной биологии. Лекция посвящена обзору нынешнего положения дел, основные проблемы воспроизводимости, их причины. Ответственность организаций, научных журналов, исследователей. Особенности проблемы в системной биологии. Основные направления разрешения проблемы воспроизводимости.
Разное
«Мотивы» – паттерны в геномных последовательностях (Иван Кулаковский, ИМБ РАН; ИОГен РАН)
Видео | Слайды
С точки зрения молекулярной биологии в лекции обсуждается регуляция активности транскрипции генов у высших эукариот и роль регуляторных белков-транскрипционных факторов. С точки зрения биоинформатики лектор рассказывает, как компьютерное представление мотивов – характерных паттернов в геномных текстах – помогает распознать регуляторные сигналы, узнаваемые транскрипционными факторами в ДНК. С точки зрения информатики рассматривает проблему построения модели 'мотива' как задачу поиска локального сходства множества последовательностей.

Аннотация промотерных последовательностей (Татьяна Татаринова, University of Southern California)
Видео | Слайды
В лекции затрагиваются вопросы закономерности и свойств промотерных последовательностей. Мотивы и метилирование промотеров. Алгоритмы предсказания и анализа промотерных последовательностей. Применение в биотехнологии.
Предсказание происхождения на основании Admixture Алгоритмы GPS и Readmix (Татьяна Татаринова, University of Southern California)
Видео | Слайды
Лекция посвящена генотипированию и отбору информативных позиций на геноме, обзору современных технологий, предсказанию био-географического происхождения человека и других организмов по анализу генома. А также анализу и сравнению существующих алгоритмов для биогеографии.
Алгоритмы в биоинформатике (Антон Банкевич, Центр алгоритмической биотехнологии, СПбГУ)
Видео | Слайды
Вводная лекция по алгоритмам в биоинформатике, в которой рассматриваются основные подходы и примеры их использования.
Связь между мозгом и Deep Learning (Дмитрий Фишман, Quretec, University of Tartu, Estonia)
Видео | Слайды
Лекция состоит из четырех частей: в первой рассматриваются пути обработки мозгом различных сигналов от внешнего мира, и формирование принятия решений на основе полученных сигналов. Во второй – эволюция методов машинного обучения, которые привели к возникновению технологии глубокого обучения (Deep Learning), осуществивших революцию во многих областях науки. В третьей части речь пойдет о сходствах и различиях между основными принципами Deep Learning. В заключении лектор приводит несколько примеров успешного применения Deep Learning в биоинформатике, и чего можно достичь в области медицинской визуализации с использованием Deep Neural Networks.
Эта лекция была создана представителями исследовательской группы по вычислительным нейронаукам Университета Тарту. В частности идея и слайды принадлежат Raul Vincente и Ilya Kuzovkin. Оригинал презентации на английском языке.

Перспективы искусственной модификации человеческих генотипов (Алексей Кондрашов, MГУ, MSU)
Видео
Никакие законы природы не запрещают синтез длинных молекул ДНК с заданной последовательностью. Каков будет фенотип человека, генотип которого не несет молодых производных аллелей? Это зависит от того, насколько распространены знаковый и сужающий эпистаз. В лекции рассматриваются рассмотрены подходы к изучению этого вопроса.
Биоинформатика в синтезе генетических конструкций (Павел Яковлев, BIOCAD)
Видео | Слайды
Развитие методов in silico молекулярного дизайна позволяет строить любые белковые конструкции с заданными свойства. Полученные аминокислотные последовательности с большой вероятностью образуют белки с нужным функционалом. Но встает новая задача: построить клеточную линию, которая бы синтезировала такие белки. В лекции рассматриваются вопросы, возникающие при решении этой задачи: почему нельзя просто взять любой обратный транскрипт, как собрать требуемый ген, как вставить его в вектор, и, конечно, причем тут биоинформатика?

Обзор современных геномных измерений отдельных клеток (Петр Харченко, Harvard University)
Видео | Слайды
Изучение сложных тканей и классификация клеточных типов традиционно базировалось на морфологических и цитологических свойствах. Несколько видов новых экспериментальных технологий теперь позволяют изучать геномные характеристики индивидуальных клеток и одновременно измерять сотни или тысячи отдельных клеток. Лекция дает обзор таких технологий и биоинформатических методов, которые используются для классификации клеточных типов, состояний и генетических линий из подобных данных.
Использование омиксных данных в изучении эволюции человека (Филип Хайтович, Shanghai Institutes for Biological Sciences, SkolTech)
Видео | Слайды
По концентрации метаболитов и липидов можно оценить физиологическое состояния тканей. В лекции представлены несколько комплексных исследований уровня концентрации метаболитов и липидов в тканях человека и животных, которые дают новые знания о молекулярных механизмах, лежащих в основе физиологических особенностей, уникальных для человека.
Послесловие
В 2016 году летнюю школу по биоинформатике поддерживали компании JetBrains, РВК, BIOCAD, EPAM Systems, Parseq Lab, за что им большое спасибо.
В 2017 году летняя школа по биоинформатике пройдет с 31 июля по 5 августа в Долгопрудном на базе МФТИ. Фокус школы в этом году – методы интеллектуального анализа данных (data mining) в биоинформатике. Дедлайн подачи заявок – 10 июня. Спешите подать заявку на участие.