Митохондриальная миопатия что это такое
Митохондриальные миопатии: причины, симптомы, диагностика, лечение
Что такое митохондриальные миопатии?
Митохондриальные заболевания вызваны нарушениями в митохондриях, которые являются источниками энергии, находящимися внутри почти всех клеток в организме. Митохондриальные заболевания, приводящие к выраженным мышечным нарушениям, называются митохондриальными миопатиями (мышцы миомы и патология - болезнь), а митохондриальные заболевания, приводящие как к выраженным мышечным, так и неврологическим нарушениям, называются митохондриальными энцефаломиопатиями («энцефало» означает, что болезнь относится к головному мозгу).
Обыкновенная клетка организма человека базируется на сотнях митохондрий для удовлетворения своих энергетических потребностей. Симптомы митохондриальной болезни различны, потому что у человека может быть уникальная смесь здоровых и дефектных митохондрий с их особым распределением в организме. В большинстве случаев митохондриальное заболевание представляет собой комплексное расстройство, влияющее более чем на один тип клеток, тканей или органов.
Поскольку мышечные и нервные клетки имеют особенно высокие энергетические потребности, мышечные и неврологические проблемы являются общими свойствами митохондриальной болезни. К другим частым осложнениям относятся нарушение зрения, сердечная аритмия, диабет и задержку роста. Как правило, у человека, страдающего митохондриальным заболеванием, имеются два или более из этих заболеваний.
Причины митохондриальных миопатий
Причиной митохондриальных заболеваний являются генетические мутации. Гены предоставляют собой наследственные инструкции для изготовления белков. А гены, порождающие митохондриальные болезни, обычно отвечают за белки, которые действуют внутри митохондрий. Внутри каждой митохондрии эти белки составляют часть сборочной линии, которая использует молекулы энергии (сахара и жиры), полученные из пищи в сочетании с кислородом, для производства молекулы аденозинтрифосфата или АТФ.
Белки в начале сборочной линии импортируют сахара и жиры в митохондрию, а затем разрушают их для обеспечения энергии. Белки в конце линии, организованной в пять групп, называемых комплексами I, II, III, IV и V, производят энергию для создания АТФ. Эта высокоэффективная часть процесса производства АТФ требует наличия кислорода и называется дыхательной цепью.
Клетка, имеющая дефектные митохондрии, лишается АТФ и может накапливать неиспользуемые молекулы топлива и разрушающие формы кислорода, называемые свободными радикалами или реактивными виды кислорода. Они, в свою очередь, контролируются антиоксидантными соединениями (обнаруженные во многих пищевых продуктах и пищевых добавках), которые обеспечивают общую защиту организма от старения и болезней.
В таких случаях молекулы избыточного топлива используются для изготовления АТФ неэффективными средствами, которые могут генерировать потенциально опасные побочные продукты, такие как молочная кислота. (Это также происходит, когда клетка имеет недостаточное снабжение кислородом, что может случиться с клетками мышц во время тяжелых упражнений.) Увеличение молочной кислоты в кровяном лактоацидозе связано с мышечной усталостью и может повредить мышечную и нервную ткань.
Мышечные и нервные клетки используют АТФ, полученный из митохондрий, в качестве основного источника энергии. Совокупные эффекты энергетической депривации и накопления токсинов в этих клетках могут приводить к различным мышечным и неврологическим симптомам.
Каковы симптомы митохондриальной миопатии?
Миопатия
Основными симптомами митохондриальной миопатии являются усталость мышц, слабость и непереносимость физических упражнений. Тяжесть любого из этих симптомов сильно различается у разных людей.
У некоторых людей слабость проявляется в мышцах, которые контролируют движения глаз и век. Двумя распространенными последствиями болезни являются постепенный паралич движений глаз, называемый прогрессирующей внешней офтальмоплегией (ПЭО), и опущением верхнего века, называемое птозом. Часто люди автоматически компенсируют ПЭО, перемещая голову, чтобы посмотреть в разных направлениях, и могут не заметить никаких проблем со зрением. Птоз может нарушить зрение, его можно исправить хирургическим путем.
Митохондриальные миопатии также могут являться причиной слабости мышц лица и шеи, что может привести к трудностям при глотании и, реже, невнятной речи. Люди с митохондриальными миопатиями также могут чувствовать мышечную слабость в руках и ногах.
Непереносимость упражнений, также называемая напряженной усталостью, относится к необычным ощущениям истощения, вызванным физическим напряжением. Степень непереносимости физических упражнений сильно различается у больных. У некоторых людей могут быть проблемы только с атлетическими упражнениями, такими как бег трусцой, в то время как другие могут испытывать проблемы с выполнением повседневных действий, например, при подъеме по лестнице.
В отдельных случаях митохондриальное заболевание связано с мышечными спазмами. В редких случаях это может привести к болям в мышцах, в частности болям после тренировки. Это нарушение приводит к вызывает утечку кислородосвязывающего белка, называемого миоглобином из мышц, в мочу (миоглобинурия). Судороги или миоглобинурия как правило возникают, когда больной «переусердствует» в отношении физических упражнений, что может произойти непосредственно во время перенапряжения или через нескольких часов после этого.
В то время как перенапряжения следует избегать, выполнение умеренных по интенсивности физических упражнение помогает людям с митохондриальной миопатией поддерживать силу мышц.
Митохондриальная энцефаломиопатия
Митохондриальная энцефаломиопатия, как правило, включает в себя отдельные симптомы миопатии и один или несколько неврологических симптомов. Опять же, эти симптомы сильно различаются у разных людей как по типу, так и по степени тяжести.
В дополнение к поражению глазных мышц митохондриальная энцефаломиопатия может воздействовать на сами глаза и части головного мозга, отвечающие за зрение. Например, потеря зрения, из-за атрофии зрительного нерва или ретинопатии, являются распространенными симптомами митохондриальной энцефаломиопатии.
Сенсибилизированная потеря слуха является распространенным симптомом митохондриальных заболеваний. Причиной чего является повреждение внутреннего уха (улитки) или слухового нерва, который соединяет внутреннее ухо с мозгом. Сенсибилизированная потеря слуха является хронической, но ее можно контролировать с помощью альтернативных форм общения, применения слуховых аппаратов или кохлеарных имплантатов. Слуховые аппараты усиливают звуки, прежде чем они достигают внутреннего уха. Кохлеарные имплантаты обходят поврежденные части внутреннего уха и стимулируют слуховой нерв.
Митохондриальные заболевания могут приводить к атаксии, которая вызывает проблемы с равновесием и координацией. Люди с атаксией подвержены падениям и могут нуждаться в использования вспомогательных устройств, таких как костыли, ходунки или инвалидные кресла.
Другими распространенными симптомы митохондриальной энцефаломиопатии являются головные боли по типу мигрени и судороги. Существует много эффективных лекарств для лечения и профилактики мигрени и судорог, включая противосудорожные препараты и другие медицинские препараты, предназначенные для лечения эпилепсии.
Особенные проблемы при митохондриальной болезни
Респираторный уход
Митохондриальные заболевания могут воздействовать на мышцы или части мозга, которые отвечают за поддержание дыхания. Больному с умеренными респираторными проблемами может потребоваться периодическая поддержка дыхания, например, с помощью дыхательных аппаратов со сжатым воздухом. Больным с более серьезными проблемами может потребоваться постоянная дыхательная поддержка в виде искусственной вентиляции легких. Необходимо следить за признаками респираторных проблем (например, одышка или утренние головные боли) и регулярно проходить обследование у специалиста по респираторным заболеваниям.
Кардиологическая помощь
Некоторые митохондриальные заболевания могут вызывать кардиомиопатию (слабость сердечной мышцы) или аритмию (нарушение ритма сердца). Аритмия поддается лечению с помощью кардиостимулятора, который стимулирует нормальное сердцебиение. Больным, страдающим митохондриальными заболевания, может потребоваться регулярное обследование кардиолога.
Другие потенциальные проблемы со здоровьем
Больные митохондриальной болезнью могут испытывать проблемы с желудочно-кишечным трактом, диабетом и / или проблемами с почками. Отдельные из этих проблем представляют собой прямые следствия митохондриальных нарушений в пищеварительной системе, поджелудочной железе (диабете) или почках, а другие – косвенные следствия митохондриальных нарушений в других тканях организма. Например, миоглобинурия нагружает почки во время фильтрации отходов крови, и может приводить к повреждению почек.
Методы лечения митохондриальных миопатий
Вместо того, чтобы сосредоточиться на конкретных осложнениях митохондриальной болезни, некоторые исследуемые методы лечения направлены на обход поврежденных митохондрий. Такие виды лечения заключаются в использовании пищевых добавок на основе трех веществ, участвующих в производстве АТФ в наших клетках.
Одно вещество, креатин, как правило, действует как резерв АТФ путем образования соединения, называемого креатинфосфатом (также называемым фосфокреатином). Когда потребность клетки в АТФ превышает количество, которое могут вырабатывать митохондрии, креатин может высвобождать фосфат («Р» в АТФ), для быстрого увеличения потребления АТФ. На самом деле, креатинфосфат обычно обеспечивает начальный всплеск АТФ, необходимый для интенсивной мышечной активности.
Другое вещество, карнитин, как правило, повышает эффективность производства АТФ, поддерживая импортирование определенных молекул энергии в митохондрии и очищая некоторые токсичные побочные продукты производства АТФ. Карнитин употребляется в форме, называемой L-карнитином.
Наконец, коэнзим Q10, также называемый CoQ10 или убихинон, является компонентом митохондриальной дыхательной цепи (которая использует кислород для производства АТФ). CoQ10 также является антиоксидантом. Некоторые митохондриальные заболевания вызваны дефицитом CoQ10, а добавка CoQ10 несомненно полезна в этих случаях и позволяет обеспечить некоторое улучшение при других митохондриальных заболеваниях.
Добавки, содержащие креатин, L-карнитин и CoQ10 часто принимаются совместно при лечении митохондриальной болезни. Несмотря на необходимость тщательных научных исследований, подтверждающих эффективность данного вида терапии, отдельные больные митохондриальным заболеванием сообщили о некотором улучшении после лечения.
Как наследуются митохондриальные миопатии?
Наследование митохондриальных заболеваний является сложным процессом, и часто митохондриальную миопатию трудно отследить через семейную ситорию. На самом деле, многие случаи митохондриальной болезни являются спорадическими, что означает, что они происходят без какой-либо наследственности.
Чтобы понять, как наследуются митохондриальные болезни, важно знать, что митохондрии связаны с двумя типами генов. Первый тип находится внутри ядра - отсека внутри наших клеток, который содержит большую часть нашего генетического материала или ДНК. Второй тип находится исключительно внутри ДНК, содержащейся внутри митохондрий.
Мутации в ядерной ДНК (nDNA) или митохондриальной ДНК (мтДНК) могут вызывать митохондриальную болезнь.
Ядерная ДНК упакована в структуры, называемые хромосомами - 22 пары хромосом, не связанных с полом (называемых аутосомами) и одной пары половых хромосом (XX у женщин и XY у мужчин). Это означает, что, за исключением генов X-хромосомы, у каждого есть две копии генов в nDNA, причем одна копия унаследована от каждого родителя. Существуют три модели наследования, наблюдаемые при заболеваниях, вызванных мутациями nDNA:
Автосомальная рецессивная способность означает, что для передачи заболевания требуется две мутантные копии гена-одного, унаследованного от каждого родителя.
Автосомальная доминантность означает, что для передачи заболевания требуется всего одна мутантная копия гена, унаследованного от одного из родителей.
Как правило, X-связанные заболевания появляются только у мужчин. Мать больного мужчины и дочери, которые у него есть, будут переносить ген болезни, но, как правило, не будут проявлять каких-либо симптомов.
В отличие от nDNA, мтДНК проходит только от матери к ребенку. Это связано с тем, что во время зачатия, когда сперма сливается с яйцом, митохондрии спермы и ее мтДНК разрушаются. Митохондриальные заболевания, вызванные мутациями мтДНК, уникальны, потому что они унаследованы по материнской схеме. Мать может передать дефектную мтДНК любому из ее детей, но только ее дочери, а не ее сыновья, передадут ее следующему поколению.
Еще одна уникальная особенность заболеваний мтДНК связана с тем, что типичная клетка организма человека содержит только одно ядро, но сотни митохондрий. Одна клетка может содержать как мутантные, так и нормальные митохондрии, а их распределение будет определять здоровье клетки, что также может объяснить диапазон симптомов при заболеваниях мтДНК.
Риск передачи митохондриальной болезни ребенку зависит от многих факторов, в том числе от того, вызвано ли это заболеванием мутациями в nDNA или мтДНК. Чтобы узнать подробнее об этих рисках, необходимо проконсультироваться с врачом или генетическим консультантом.
Диагностика митохондриальных заболеваний
Характерными признаками митохондриальной миопатии являются мышечная слабость, непереносимость физической нагрузки, нарушение слуха и зрения, атаксия, судороги, нарушения зрения, сердечные дефекты, диабет и плохой рост - ни одна из которых не является характерной только для митохондриальной болезни. Однако сочетание трех или более этих симптомов у одного человека в высокой степени свидетельствует на митохондриальную болезнь, особенно когда симптомы связаны с более чем одной системой органов.
Для оценки характера данных симптомов, врачу необходимо изучить историю болезни человека. Поскольку митохондриальные заболевания являются генетическими, семейная история также является важной частью постановки диагноза. Для диагностики также проводятся физические и неврологические исследования и тесты.
Физические тесты обычно включает в себя тесты на силу и выносливость, например, физические упражнения (которые может включать в себя такие действия, как повторяющееся сжимание руки в кулак). Неврологические тесты могут включать в себя тесты на рефлексы, зрение, речь и основные познавательные навыки.
Как правило, врач назначает лабораторные анализы на диабет, проверку печени и почек. Врачом, скорее всего, также назначается электрокардиограмма (ЭКГ), для проверки сердца на наличие признаков аритмии и кардиомиопатии.
Исследования могут назначаться для проверки наличия патологий в головном мозге и мышцах. Диагностическая визуализация, позволяющая получить подробные снимки органов, костей и тканей, а именно, компьютерная томография (КТ) или магнитно-резонансная томография (МРТ), может использоваться для проверки головного мозга на нарушения развития или признаки повреждения. Больному, у которого случаются приступы, врачом может назначаться электроэнцефалограмма (ЭЭГ), заключающаяся в размещении электродов на голове для регистрации эектрической активности мозга.
Так как лактоацидоз является общей чертой митохондриального заболевания, то, как правило, проводится анализ для проверки на повышенный уровень молочной кислоты в крови и моче. Некоторые случаи могут потребовать измерения молочной кислоты в спинномозговой жидкости (CSF), которая заполняет пространства в мозге и спинном мозге. Измерение может быть выполнено путем забора спинномозговой пункции.
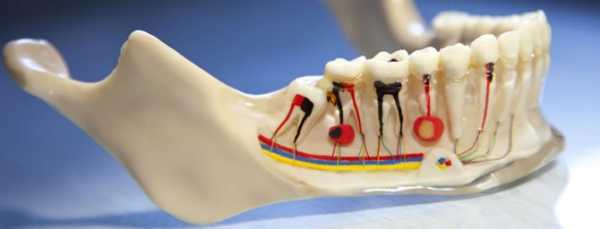
Одним из наиболее важных исследований при диагностике митохондриальной болезни является биопсия мышц, которая заключается в исследовании небольшого образца мышечной ткани. При введении красителя, окрашивающим митохондрию красным цветом, мышцы, пораженные митохондриальной болезнью, часто показывают рваные красные волокна - мышечные клетки (волокна), имеющие чрезмерное количество митохондрий. Другие пятна могут обнаружить отсутствие существенных митохондриальных ферментов в мышцах. Также возможно извлечение митохондриальных белков из мышцы и измерение их активности.
Неинвазивные методы могут быть использованы для исследования мышц без взятия образца ткани. Например, МР спектроскопия может использоваться для измерения уровней органической молекулы фосфокреатина и АТФ (которые часто истощаются в мышцах, пораженных митохондриальным заболеванием).
Наконец, генетическое тестирование может определить, имеет ли кто-то из родственников больного генетические мутации, которые вызывают митохондриальную болезнь. Эти тесты используют генетический материал, извлеченный из крови или из биопсии мышц. Хотя положительный результат теста может подтвердить диагноз митохондриального расстройства, отрицательный результат теста может быть сложно интерпретировать. Что может означать, что у человека есть генетическая мутация, которую тест не смог обнаружить.
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕЕЩЕ: НА ГЛАВНУЮ СТРАНИЦУ
Митохондриальные миопатии
Перевод материалов сайта Muscular Dystrophy UK
Ссылка на оригинал, а также сохранённый pdf-файл с оригиналом статьи смотреть в конце.
Что такое митохондриальная миопатия?
Митохондриальная миопатия – термин, объединяющий группу заболеваний, которые в большинстве случаев поражают мышцы, но также могут затрагивать и любую другую часть тела, включая мозг и глаза. Это заболевание имеет и другие названия: cиндром Кернс-Сейр (KSS), хроническая прогрессирующая внешняя офтальмоплегия (CPEO), синдром MELAS, миоклоническая эпилепсия с рваными мышечными волокнами (MERRF), синдром Лея и митохондриальная цистопатия.
Почему возникает это заболевание?
Наши тела состоят из множества различных тканей, к примеру, мышечной, нервной или печеночной. Каждая ткань построена из маленьких строительных блоков, клеток. Каждая клетка имеет митохондрии. Их роль заключается в производстве энергии. Как и любой электрогенератор, они берут топливо (еду, что мы потребляем) и сжигают его, чтобы произвести энергию. Если этот процесс нарушается, клетки не могут нормально функционировать. Это и приводит к болезни. Мышцам и мозгу нужно много энергии, и при её нехватке именно эти органы “страдают” больше всего.
Часть митохондрии, задействованная в производстве энергии, называется дыхательной цепью. Компоненты (белки) этой цепи производятся в соответствии с генетическим кодом (ДНК) и находятся либо внутри самих митохондрий (мтДНК), либо в хромосомах, которые расположены в ядрах клетки. Многие митохондриальные болезни носят спорадический характер, даже если они затрагивают мтДНК. Это значит, что если один член семьи имеет данную болезнь, его родители или дети могут и не иметь данного заболевания. Другие митохондриальные заболевания наследуются только по материнской линии. Те же заболевания, которые возникли из-за дефекта в хромосомах, могут быть унаследованы от любого из родителей.
Как выявляют болезнь?
Если при обследовании обнаружены такие симптомы как опущение верхнего века (птоз) или нарушения движения глазных яблок (офтальмоплегия), можно заподозрить митохондриальную миопатию. Независимой организацией Muscular Dystrophy Campaign (MDC) составлен список симптомов миопатии глаз. Анализ крови может показать повышенный уровень молочной кислоты. Окончательный диагноз часто зависит от результатов проб мышечной биопсии, для которых доктор берёт небольшой фрагмент мышечной ткани для лабораторных исследований. Под микроскопом митохондрии у людей с митохондриальной миопатией часто отличаются от нормальных. Митохондрии собираются по краям мускульной ткани, придавая ей рваный, красноватый вид. Также возможно измерить функциональность дыхательной цепи и установить дефектную зону. В некоторых случаях возможно поставить диагноз по дефекту (мутации) в генетическом коде (в мтДНК или клеточном ДНК).
Что происходит с людьми, болеющими митохондриальной миопатией?
Митохондриальная миопатия может проявляться по-разному. Самой частой проблемой являются совокупность таких симптомов как слабость рук и ног с опущением верхнего века (птоз) и нарушение двигательной функции глаз (офтальмоплегия). У других пациентов заболевание может проявляться только в слабости рук и ног и может усиливаться после нагрузок. Болезнь может сопровождаться тошнотой и головной болью. При тяжёлой форме болезни мышечная слабость может быть заметна уже у младенцев. Им может быть тяжело глотать и есть. Реже поражается мозг. Это может вызывать эпилепсию (приступы) и прогрессивную потерю памяти. Лишь у некоторых пациентов с поражённым мозгом (энцефалопатия) наблюдаются дальнейшие ухудшения. Часто поражается сетчатка глаза (нарастает атипичный пигмент), и возникают проблемы со слухом. В случае поражения сердца требуется установка кардиостимулятора – электронного устройства, нормализующего работу сердца.
Насколько опасны заболевания и каков прогноз?
Самый часто встречающийся вид митохондриальной миопатиии – хроническая прогрессирующая внешняя офтальмоплегия (CPEO). Это небольшое нарушение, которое не отражается на продолжительности жизни. Однако другие виды, которые проявляются на раннем этапе, представляют угрозу. Некоторые, такие как наследственная оптическая нейропатия Лебера (LHON), влияют только на зрение. Эпилепсия, проблемы с сердцем и трудности с дыханием представляют большую угрозу.
Существует ли лечение и возможно ли полное восстановление?
Не существует чудодейственных лекарств. Однако многие проблемы, связанные с митохондриальной миопатией, могут быть успешно решены. К примеру, диабет можно лечить таблетками или инсулином. Кардиостимуляторы эффективно справляются с нарушениями сердечного ритма. Мышечную слабость можно контролировать регулярными лёгкими упражнениями. Некоторые пациенты улучшают своё состояние здоровья с помощью витаминов, таких как убихинон, но большинству витаминотерапия не помогает.
С какими особыми трудностями придётся столкнуться?
Когда мы болеем, потребность в энергии возрастает. Добровольный или лечебный пост также увеличивает нагрузку на митохондрии. Поэтому важно регулярно потреблять калории в течение дня. Если в этом возникают сложности, в особенности во время болезни или если пациент − маленький ребёнок, рекомендуется обратиться к врачу. Некоторые препараты могут повлиять на функционирование митохондрий. Обычно советуют избегать алкоголя. Если возникают сомнения, следует посоветоваться со специалистом.
Опасно ли это для членов моей семьи?
В результате недавних исследований была получена информация о генетическом аспекте митохондриальной миопатии. Можно утверждать, что риски для других членов семьи зависят от точного диагноза. У большинства людей с митохондриальной миопатией нет родственников с похожим заболеванием, однако такое случается только в 20% случаев. Это заболевание может быть наследственным (передаётся от родителя к ребёнку) или произойти в результате ошибки в генетическом коде (мутации) ДНК самой митохондрии или ядра клетки. У людей с дефектами в мтДНК болезнь наследуется от матери. У людей с дефектом в ядрах клеток заболевание может быть получено от любого из родителей.
Что если я захочу ребёнка? Каков шанс, что я не передам дефектный ген ребёнку?
Это сложный вопрос. Ответ будет зависеть и от пациента, и от типа заболевания (к примеру, от кого из родителя передан ген). В случае, если болезнь передаётся от матери, использование яичников (ооцитов) незаражённого донора сводит риск передачи к нулю. Хотя, конечно, донором не должен являться родственник по материнской линии (к примеру, сестра). При определённых видах митохондриальной миопатии возможно исследовать плаценту младенца (CVS) и даже эмбриона на ранней стадии (предимплатанционый диагноз). Однако зачастую такая процедура либо почти недоступна, либо неприменима к определённым видам нарушений.
Местная генетическая клиника сможет дать дальнейший совет. В случае с синдромом Лея, если мутацию обнаружили у ребёнка, возможно поставить диагноз до родов и получить помощь специализированных центров.
Что делать, если я уже беременна?
Во время беременности сложно оценить, насколько сильно болезнь повлияет на ребёнка.
Чем я могу помочь себе (своему ребёнку)?
Важно придерживаться сбалансированного питания, включая адекватный приём витаминов и недопущения ожирения. Так как пост увеличит нагрузку на митохондрии, рекомендован регулярный приём пищи с высоким содержанием углеводов, если есть возможность. Следует избегать чрезмерных физических нагрузок. Их уровень будет зависеть от того, насколько тяжело повреждены мышцы. Но для тех, кто может выполнять физические нагрузки, спорт улучшит самочувствие, а в некоторых случаях может улучшить функционирование мышц.
Специальные учреждения могут предоставить практическую помощь и информацию. Они также помогают установить связи между различными профессионалами, такими как физиотерапевты, профпатологи, социальные работники и учителя.
[Напоминаем, что статья от лица Muscular Dystrophy Campaign, поэтому помощь возможна на территории Соединённого Королевства. Однако, вполне вероятно, что эта организация помогает людям и из других стран. Прим. администраторов сайта]Мы будем с вами на момент постановки диагноза и на всем дальнейшем пути. Мы сможем:
- дать вам самую новую и точную информацию о вашем состоянии и состоянии вашего ребёнка;
- дать вам знать о прогрессе в исследованиях, снабдим вас советами на каждый день, написанные людьми, которые знают, каково это жить с мышечной атрофией;
- познакомить с семьями, которые живут с таким же заболеванием и которые смогут поделиться с вами опытом;
- поделиться информацией, помочь вам, услугами, оборудованием и поддержкой, на которые вы имеете полное право.
Если вам бы хотелось, чтобы ваш врач имел больше информации о митохондриальной миопатии, у нас есть подходящие материалы. Мы разработали онлайн-обучение для врачей, работающих со взрослыми, имеющими атрофию мышц. Свяжитесь с нами по горячей линии или по телефону, чтобы получить больше информации.
Отказ от прав: Несмотря на то, что мы делаем всё возможное, чтобы удостоверится, что информация в этом документе полна, верная и соответствует последним исследованиям, не можем это полностью гарантировать. Организация Muscular Dystrophy UK не несёт ответственности в случае причинении какого-либо вреда в результате использования данной информации. Организация Muscular Dystrophy UK не обязана поддерживать услуги, которые предоставляются организациями в списке.
Оригинал: Mitochondrial myopathies
http://www.musculardystrophyuk.org/app/uploads/2015/02/Mitochondrial-myopathies-2015.pdf
Перевод: Екатерина Глухова
Митохондриальная миопатия: причины, симптомы, диагностика, лечение
Митохондриальная миопатия – это термин, который объединяет группу заболеваний, в большинстве своем поражающих мышцы (реже – глаза, мозг и другие части тела).
Какие факторы провоцируют развитие подобных патологий? Каковы причины? Какие симптомы указывают на наличие у человека миопатии? И, самое главное, как лечить такое состояние? Об этом и многом другом сейчас речь и пойдет.
Типы митохондриальных заболеваний
С их рассмотрения хотелось бы начать. Каждый недуг данного характера обусловлен биохимическими, структурными или генетическими дефектами митохондрий. Они приводят к одному и тому же последствию – к нарушению тканевого дыхания.
Передается предрасположенность исключительно по женской линии, но к детям обоих полов. Патологические нарушения обычно проявляются в дефектах разных звеньев в дыхательной цепи, цикле Кребса или процессах бета-окисления.
Стоит оговориться, что далеко не все регуляторы и ферменты, являющиеся необходимыми для полноценного функционирования митохондрий, проходят кодировку через митохондриальную ДНК. В ряде случаев в данном процессе принимает участие ядерная ДНК.
И перед тем как перейти к подробному рассказу о том, что это такое – митохондриальная миопатия, следует рассмотреть группы заболеваний данного происхождения. Всего их две:
- Наследственные синдромы, которые обусловлены генными мутациями. Это синдромы Кернс-Сейра, Пирсона, Барта, MELAS, MERRF и т. д.
- Вторичные заболевания, которые включают нарушение клеточного энергообмена. Сюда относится гликогеноз, печеночная недостаточность, рахит, диабет, гипопаратиреоз, панцитопения, мигрень, а также синдром хронической усталости.
Типов митохондриальных наследственных заболеваний существует намного больше. В список включены такие недуги:
- Сахарный диабет, сопровождающийся глухотой.
- Синдром Вольфа-Паркинсона-Уайта. Сердечная патология, характеризующаяся аномальным возбуждением желудочков.
- Оптическая нейропатия Лебера. Практически всегда чревата потерей зрения, которая приходится на ранний пубертатный период.
- Энцефалопатия нейрогастроинтенстинального характера.
- Рассеянный склероз.
- Атаксия, нейропатия.
- Туннельное зрение, слепота, деменция, птоз.
- Синдром Лея. Также известен как подострая некротизирующая энцефаломиопатия.
Следует оговориться, что изначально мутации такого рода считались редкими. Но в результате исследования на 10 известных генетических аномалий, в котором принимало участие 3000 новорожденных, таковые обнаружились в довольно большом количестве – у 1-го младенца из 200.
Этиология
Митохондриальная миопатия формируется вследствие нарушений, возникающих в митохондриях. Они являются источниками энергии, которые находятся внутри практически всех клеток организма.
Каков патогенез? Так называемой базой обыкновенной клетки являются сотни митохондрий. Их общей энергии ей хватает для развития и поддержания жизнедеятельности.
Однако некоторые митохондрии могут быть дефектными. Из-за их наличия клетка лишается аденозинтрифосфата – вещества, имеющего огромное значение в процессе обмена веществ. Следствием становится накапливание излишнего количества молекул топлива и разрушающих форм кислорода.
Почему же некоторые митохондрии оказываются мутированными? Причина заключается в генетических мутациях. Ведь гены – это наследственные инструкции. Они задействованы в процессе «изготовления» белков. А те гены, которые порождают патологии указанного характера, отвечают за белки, действующие именно внутри митохондрий.
Патогенез
Что же происходит вследствие вышеописанного? Организм начинает использовать молекулы избыточного топлива, хотя нужды в этом нет. В результате генерируются побочные продукты, являющиеся потенциально опасными (молочная кислота, например). То же самое происходит в тех случаях, когда клетке не хватает кислорода. Такое, кстати, случается и в момент выполнения тяжелых упражнений.
Что в итоге? Уровень молочной кислоты в кровяном лактоацидозе увеличивается. Из-за этого возникает мышечная усталость. И данное явление часто становится причиной повреждения мышечной и нервной ткани.

Иначе говоря, совокупные эффекты энергетической депривации, а также накопления в клетках токсинов, приводят к появлению неврологических и мышечных симптомов. Последние и указывают на сформированную митохондриальную миопатию.
Симптомы
Теперь следует поговорить о признаках, по которым можно узнать о наличии заболевания, относящегося к рассматриваемой группе патологий.
Ключевые симптомы митохондриальной миопатии выделяют в такой перечень:
- Сильная усталость мышц.
- Непереносимость физических упражнений, даже не требующих особой нагрузки.
- Слабость.
- Судороги и парезы.
- Одышка.
- Изменение цвета мочи.
- Головные боли, которые по характеру напоминают мигрень.
Поражаются мышцы конечностей, часто диагностируется повреждение волокон глазного яблока (это проявляется офтальмоплегией и птозом). В патологический процесс может вовлекаться ЦНС, в таких случаях диагностируют деменцию, миоклонии, эпилепсию, мозжечковую атаксию и т. п.
Нередко поражение распространяется на почки (тубулопатия), сердце (кардиомиопатия), слуховой анализатор, печень.
Еще важно оговориться, что обычно возникает митохондриальная миопатия у детей. Однако «дебютировать» патология может и в старшем возрасте.
Как бы там ни было, мышечная слабость имеет обычно распространенный характер, по которому большинство пациентов и узнают о своем недуге. Реже встречаются ограниченные проявления – локальные поражения мышц лица, конечностей, глазного яблока.
Диагностика
Поговорив о симптомах митохондриальной миопатии, можно перейти к изучению особенностей диагностики. Обычно подозрение данной патологии у врачей возникает после обнаружения птоза (опущение верхнего века) либо нарушения движений глазных яблок.
В таких случаях пациента направляют на сдачу крови на анализ. Результаты, как правило, демонстрируют увеличенный уровень молочной кислоты. Но это тоже не может быть основанием для постановки точного диагноза. Все сомнения развеять способны результаты пробы мышечной биопсии.
Изучив в лабораторных условиях фрагмент мышечной ткани, можно обнаружить многое. Митохондрии людей с данной патологией отличаются от здоровых, не мутированных. Они собираются на краях мускульной ткани, из-за чего та обретает красноватый, рваный вид.

Еще часто назначают измерение функциональности дыхательной цепи. Это нужно для установления дефектной зоны. И лишь в некоторых случаях удается поставить диагноз по мутации в генетическом коде.
Вообще, без сбора полной анамнестической картины и подробного клинического обследования диагностировать митохондриальную миопатию у детей или взрослых невозможно. Основная сложность заключается в отсутствии жалоб у пациентов в состоянии покоя. Поэтому ряд показанных диагностических процедур велик. Он включает в себя:
- Исследование ферментативной активности (сдача крови на ферменты ЛДГ, КФК, АЛТ, АСТ, адолазы и т. д.).
- Электронейрография (альтернатива – электромиография).
- Ишемический тест на предплечье, необходимый для выявления гликогеноза.
- Томография внутренних органов.
- КТ или МРТ головного мозга.
- Биопсия мышц с микроскопическим, гистохимическим, а также морфологическим исследованием.
Кроме всего перечисленного, для точной диагностики митохондриальной миопатии часто требуется пройти консультацию нефролога, кардиолога, невролога, генетика и офтальмолога.
Осложнения
Митохондриальная миопатия является очень тяжелым нарушением, чреватым серьезными последствиями. Почему важно не пренебрегать терапией, даже если прогноз малоприятный? Потому что если запустить заболевание, можно допустить развитие осложнений. К ним относятся такие состояния:
- Потеря слуха.
- Деменция.
- Лактоацидоз.
- Резкое снижение веса.
- Миоклоническая эпилепсия прогрессивного типа.
- Ограниченная подвижность глаз.
- Нарушения нервной системы.
- Рвота.
- Скелетные аномалии мышц.
- Расстройства ЖКТ.
- Затрудненность в глотании.
- Сердечная недостаточность или нарушенный ритм.
- Судороги.
- Слабоумие.
И это далеко не все возможные последствия митохондриальной миопатии. Дают ли инвалидность при таком состоянии? Логичный вопрос. Поскольку данная патология накладывает весомый отпечаток на работоспособность человека, то да. Заболевание с поражением нервно-мышечного аппарата, подтвержденное медицинским учреждением – основание для получения инвалидности.
Терапия
Лечение митохондриальной миопатии, к сожалению, не представляется возможным. Потому что проблема заключается во врожденном нарушении обмена веществ. Говоря простым языком, в системе имеется сбой, который устранить нереально.
Однако без терапии не обойтись никак. Симптоматическая помощь должна быть. То, какие препараты придется принимать пациенту, определяет врач после проведения диагностики митохондриальных заболеваний.

Также неотъемлемой составляющей жизни пациента становится диетотерапия. Очень важно употреблять продукты с повышенным содержанием фруктозы и белка. Сочетая такой рацион с поливитаминами, можно улучшить состояние, особенно при гликогенозах.
Если же у пациента диагностирована миоглобинурия, то назначают введение в организм дополнительного количества жидкости. Это необходимо для профилактики почечной недостаточности и диуреза.
И в обязательном порядке при митохондриальной миопатии назначают прием медикаментов, цель которых – улучшение энергетического метаболизма. Это рибофлавин, L-карнитин, витамины Е и С, тиамин и убихинон.
Важно оговориться, что отличные результаты показывает употребление глюкокортикостероидов. Еще часто назначают ортопедическую коррекцию.
Отдельными случаями являются пациенты с приобретенными и вторичными миопатиями. Здесь терапию направляю на устранение причины митохондриальных заболеваний – корректируют системные недуги, эндокринные патологии, интоксикации и т. д. Говоря простым языком, в таких случаях лечат то заболевание, которое и спровоцировало появление миопатии. Уйдет оно – устранится и данное последствие.
Физкультура и массаж
Это две ключевые составляющие терапии при миопатии. Человеку, страдающему данным недугом, нельзя вести малоподвижный или сидячий образ жизни. Это лишь усугубит его состояния.
А вот лечебная физкультура позволяет предотвратить атрофию мышц и дальнейшее развитие контрактуры. Приятным бонусом является улучшение мышечного тонуса.
Особенно важно вовремя начать такое лечение при миопатии Дюшенна. Полезно выполнение упражнений в воде (в бассейне, например, а лучше в море), стоит делать акцент на конечности.
Обычно пациенту назначают индивидуальный курс лечебной физкультуры. Нагрузка, разумеется, минимальная – посильная для пациента. Когда человек пройдет курс, то останавливаться не рекомендуется. Наоборот, нужно ежедневно выполнять упражнения в домашних условиях, хотя бы немного усиливая нагрузки.
Если с миопатией столкнулся совсем маленький ребенок, то ему нужно активно поддерживать тонус голеностопных мышц, поскольку именно этот отдел влияет на качество ходьбы.

Это нормально, когда у человека не получается справляться с физкультурой. А потому поначалу дозировка общеразвивающих упражнений минимальна – 4 раза для каждого, не более. И использовать подручные средства для их выполнения разрешено.
Что касается массажа, он избирательный, специалист интенсивнее воздействует на наиболее пораженные мышцы. Рекомендованный курс – 1,5 месяца с ежедневными сеансами.
Помимо перечисленного, настоятельно рекомендуется ходить на занятия в бассейн как минимум 2 раза в неделю. Еще позитивно на состояние здоровья отражаются упражнения на дыхание.
Что еще нужно знать?
Когда речь идет о миопатии, очень важно осознать – лечение генетической аномалии либо наследственного генного заболевания, пока не разработано. Единственный вариант – симптоматическая терапия, способная улучшить качество жизни. Ее цель – восстановить и усилить происходящие в мышечных тканях метаболические процессы.
Обязательно употребление витаминов группы В. Они широко известны своим антиневритным действием. Именно эти препараты способны усилить, а затем и восстановить нервно-мышечные импульсы в поврежденных структурах. Но принимать их нужно лишь параллельно с аденозинтрифосфорной кислотой. Она является элементом, который модулирует энергетические обменные реакции на клеточном уровне.

Еще терапия подразумевает прием антихолинэстеразных средств. К таковым относится галантамин и амбеноний. Они оказывают целый ряд положительных действий:
- Накопление ацетилхолина.
- Усиление выносливости.
- Улучшение нервно-мышечной передачи.
- Повышение активности.
Кроме этого, к приему показана глютаминовая кислота и гидролизаты. Употребление этих средств способствует восполнению запасов организма, необходимых для построения пресловутых мышечных волокон.
И, разумеется, не обходится без употребления микроэлементов. Говоря простым языком, медикаментозная терапия является многосоставной, комбинированной. Длительность колеблется от одного до двух месяцев, но за год пациент проходит как минимум три таких курса.
Диета
Раз речь идет об особенностях митохондриальных заболеваний и их лечении, то нужно в отдельности поговорить о рационе, показанном к соблюдению людям, столкнувшимся с миопатией.
Главная цель диеты – улучшить здоровье, перестроить обмен веществ, а также заменить главное «топливо» организма на жиры.
Каковы основные положения рациона? В нем должны содержаться высококачественные жиры в большом количестве, малый объем углеводов и умеренный – протеина. Соблюдение такой диеты направлено на оптимизацию способности митохондрий вырабатывать кетоны – органические вещества, лучше всего подходящие на «роль» основного топлива организма.
Однако чтобы организм начал сжигать их, надо увеличить количество употребляемых здоровых жиров и свести к минимуму объем съедаемых углеводов.
Прогноз
Выше было многое сказано о митохондриальной миопатии. Что это – ясно. Каковы же прогнозы? К сожалению, неблагоприятные. Особенно когда речь идет о младенческих формах и тех заболеваниях, которые дают о себе знать с самых ранних недель жизни ребенка. Такие пациенты часто погибают в начале новорожденного периода.

У поздних и взрослых форм прогноз более благоприятный. Но то, как будет протекать патологический процесс, а также каков будет итог, зависит еще и от вовлечения в него других систем и органов. Если, например, поражено еще сердце, почки и печень, то прогноз будет также неблагоприятным.
Шансы на улучшение качества жизни имеют пациенты с приобретенной митохондриальной миопатией. Это не может не радовать. Симптомы их патологического процесса удается смягчить, проведя коррекцию причинного заболевания.
Необходимо оговориться, что нельзя пренебрегать профилактикой первичных метаболических патологий. В чем же она заключается? В генетическом медицинском консультировании мужчины и женщины, решившихся на зачатие. Его нужно будет провести перед планированием беременности, а потом еще и в первом триместре.
Для чего это нужно? Своевременное выявление пресловутых вторичных миопатий позволяет быстро начать терапию, благодаря проведению которой можно избежать развития нарушений в мышечной ткани.
Митохондриальные миопатии и энцефаломиопатии | МКДЦ ФГБНУ НЦН
Митохондриальные миопатии и энцефаломиопатии — группа заболеваний, обусловленных генетическими дефектами митохондрий, сопровождающимися нарушением тканевого дыхания.
КЛИНИЧЕСКАЯ КАРТИНА
Для митохондриальных миопатий характерна полиорганнасть патологии, относительная динамичность симптомов, сочетание с эпилептическими приступами, инсультоподобными эпизодами, пигментным ретинитом, мозжечковой атаксией, нейросенсорной тугоухостью, нарушением проводимости сердца и другими симптомами. Выраженность клинической симптоматики варьирует от субклинических изменений до тяжёлых фатальных случаев, что связано с уровнем гетероплазмии и выраженностью митохондриального дефекта. Синдром MERRF характеризуется сочетанием миопатии, миоклонии (60%), эпилептических приступав (45%), атаксии, деменции, атрофии зрительных нервов (20% случаев) и тугоухости. В 20% случаев наблюдают полиневропатию (сенсорные нарушения). MELAS характеризуется началом в детском возрасте, низкорослостью, инсультоподобными эпизодами (85%) , многократными приступами рвоты (90%), тугоухостью (25%), миоклонической эпилепсией, деменцией (50% больных), умеренной проксимальной миопатией, хронической прогрессирующей наружной офтальмоплегией. Для синдрома Кирнса-Сейра характерна хроническая прогрессирующая наружная офтальмоплегия, проксимальная мышечная слабость (90%), дисфагия (50%), нарушение проводимости сердца, атаксия (90%), пигментная ретинопатия, тугоухость (90% случаев). Также существует изолированная хроническая наружная прогрессирующая офтальмоплегия, связанная с митохондриальной патологией, которая обычно начинается в зрелом возрасте.
СИМПТОМЫ
Для синдрома Кирнса-Сейра характерна хроническая прогрессирующая наружная офтальмоплегия, проксимальная мышечная слабость (90%), дисфагия (50%), нарушение проводимости сердца, атаксия (90%), пигментная ретинопатия, тугоухость (90% случаев) . Также существует изолированная хроническая наружная прогрессирующая офтальмоплегия, связанная с митохондриальной патологией, которая обычно начинается в зрелом возрасте. Возможны птоз различной степени выраженности, чаще двусторонний (асимметричный или симметричный), наружный офтальмопарез (ограничение подвижности глазных яблок, не укладывающееся в топику поражения глазных нервов, диплопия, более выраженная в крайних отведениях глазных яблок), слабость мимической мускулатуры. Мышечная слабость умеренная, больше выражена в проксимальных отделах конечностей. Сухожильные рефлексы часто сохранны. Могут развиться контрактуры ахилловых сухожилий.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. Немедикаментозное лечение- рекомендуют общее для всех миопатий лечение: ЛФК, массаж. Медикаментозная терапия включает энерготропные препараты, витамины, антиоксиданты.
признаки, симптомы, диагностика и лечение

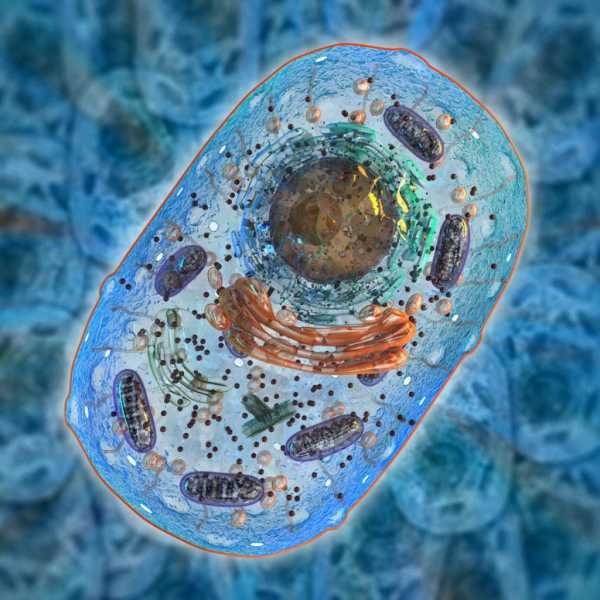
Митохондрии - структуры внутри клеток тела, которые преобразуют кислород и вещества из пищи в энергию. Их можно рассматривать как единственный источник энергии клеток. Эти «электростанции» обеспечивают поступление энергии к мышцам и головному мозгу.
Существует ряд заболеваний, вызванных сбоями митохондрий, имеющих общий термин «митохондриальные болезни». Причиной этих заболеваний является неспособность митохондрии генерировать достаточное количество энергии, необходимое для нормального функционирования клетки.
Митохондриальная миопатия - расстройство, относящееся к группе нейро-мышечных заболеваний, которые не только влияют на мышцы, но и на все остальные части тела, включая мозг и глаза. Нервные клетки в головном мозге и мышцах требуют больше энергии, и, таким образом, получают особые повреждения при возникновении митохондриальной дисфункции. Это наследственное заболевание.
Симптомы хромосомной миопатии.
Данное заболевание возникает в возрасте до 20 лет, и часто начинаются с непереносимости физической нагрузки или мышечной слабости. Во время физической активности, мышцы могут легко стать усталыми или слабыми, однако мышечные спазмы происходят редко. Может наблюдаться тошнота, головная боль и одышка, а также:
• лактоацидоз;• потеря слуха;
• потеря веса;
• различные типы деменции;
• прогрессивная миоклоническая эпилепсия;
• висящие веки и ограниченная подвижность глаз;
• рвота;
• нарушения нервной системы;
• проблемы со зрением;
• скелетные аномалии мышц;
• мышечная слабость и двигательные расстройства;
• расстройства желудочно-кишечного тракта;
• усталость и непереносимость физической нагрузки;
• трудность глотания;
• судороги;
• нарушения ритма или сердечная недостаточность ;
• слабоумие.
Причины хромосомной миопатии.
Заболевание в основном возникает из-за мутаций в митохондриальной ДНК или в ядерных генах, имея приобретенный или наследственный характер. Хромосомная миопатия также может возникнуть из-за митохондриальной дисфункции, вызванной в результате побочных эффектов от лекарств, окружающей среды или инфекций. Во время клеточного деления, митохондриальная ДНК случайным образом разделяется между двумя новыми митохондриями, полученными от отца и матери, создавая несколько копий. Митохондриальная болезнь становится клинически очевидной, когда число пострадавших митохондрий, унаследованных от матери, увеличивается. Дефекты ферментов, которые контролируют митохондриальной репликации ДНК, также могут вызывать мутации митохондриальной ДНК. Основная часть митохондриальной функции и биогенез контролируется ядерной ДНК. Сбои в ядерных кодировках митохондриальных генов связаны с сотнями клинических фенотипов заболеваний, в том числе малокровием, ретинопатией, лимфомой, повышенным кровяным давлением, слабоумием.
Диагностика и лечение митохондриальной миопатии.
Диагностика заболевания включает в себя клиническое наблюдение, лабораторные анализы и генетические исследования. Перед установкой диагноза, тщательно изучается история болезни пациента.
Физический осмотр может выявить некоторые особенности митохондриальной миопатии, например висящие веки (птоз), или трудность перемещения глаз (офтальмоплегия). Для выявления повышенного уровня ферментов и наличие белка, делается общий анализ крови.
Окончательный диагноз часто зависит от результата биопсии мышечной ткани (удаляется небольшой кусок мышцы для дальнейших лабораторных испытаний). Под микроскопом изучается вид, функционирование и наличие аномалий митохондрий. В некоторых случаях можно установить диагноз путем поиска ошибки в ДНК. При помощи генетического анализа крови можно выделить мутированные гены. С целью диагностики жидкости, которая окружает спинной и головной мозг проводится люмбальная пункция.
Лечение исключительно направлено на облегчение боли, симптомов заболевания и восстановление максимальной подвижности, и чаще всего проводится неврологом или генетическим специалистом.
Варианты лечения могут включать в себя:
• Физиотерапию (данный метод лечения проводится для укрепления и улучшения движения мышц). В некоторых случаях для поддержки движения могут быть использованы фигурные скобки и инвалидные коляски.• Дыхательную терапию (использование искусственной вентиляции легких).
• Логопедию (пациенты, страдающие данным заболеванием, часто имеют проблемы с речью, что требует речевой терапии).
• Пищевые добавки (креатин, коэнзим Q10, карнитин, липоевая кислота, рибофлавин, витамин К или идебенон) могут помочь увеличить энергию в клетках.
• Лекарственные препараты используются для лечения специфических симптомов, таких как судороги и боли.
Продолжительность митохондриальной миопатии может быть различной и часто зависит от типа заболевания и степени вовлечения различных органов.
Митохондриальные заболевания — Википедия
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК[1].
Можно выделить две группы митохондриальных заболеваний:
- Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
- Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Наследование митохондриальных болезней[править | править код]
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все митохондрии наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между её потомками. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеванийДефекты и симптомы[править | править код]
Эффекты митохондриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае митохондриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани,[2] поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Помимо относительно распространённой митохондриальной миопатии, встречаются:
- митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
- наследственная оптическая нейропатия Лебера (en:Leber's hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде;
- синдром Вольфа-Паркинсона-Уайта (en:Wolff-Parkinson-White syndrome) Синдром WPW не относится к митохондриальным миопатиям.
- рассеянный склероз и подобные ему заболевания;[источник не указан 3038 дней]
- синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в конце первого года жизни, иногда — во взрослом возрасте. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
- нейропатия, атаксия, retinitis pigmentos и птоз en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, туннельное зрение и потеря зрения, птоз, деменция;
- митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200.[3] «Горячей точкой» в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов[4]. Также в качестве одного из методов применяются пируваты[5].
В настоящее время проводятся экспериментальные работы по изучению возможности экстракорпорального (in vitro) оплодотворения с использованием химерной яйцеклетки, ядро которой получено из яйцеклетки пациентки с митохондриальным заболеванием, а цитоплазму из другой яйцеклетки от женщины с нормально функционирующими митохондриями (замена ядра)[6].
- ↑ Scarpulla R.C. Nuclear control of respiratory gene expression in mammalian cells (англ.) // J Cell Biochem. : journal. — 2006. — Vol. 97, no. 4. — P. 673—683. — PMID 16329141.
- ↑ Finsterer J. Hematological manifestations of primary mitochondrial disorders (англ.) // Acta Haematol. : journal. — 2007. — Vol. 118, no. 2. — P. 88—98. — doi:10.1159/000105676. — PMID 17637511.
- ↑ Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2008. — August (vol. 83, no. 2). — P. 254—260. — doi:10.1016/j.ajhg.2008.07.004. — PMID 18674747.
- ↑ «When Cells Stop Working» dated November 5, 2006 at Time Magazine
- ↑ Tanaka M., Nishigaki Y., Fuku N., Ibi T., Sahashi K., Koga Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases (англ.) : journal. — 2007. — doi:10.1016/j.mito.2007.07.002. — PMID 17881297.
- ↑ Hello mothers, hello father (англ.). The Economist (27 October 2012). Дата обращения 17 декабря 2013.
На русском языке[править | править код]
На английском языке[править | править код]
Митохондриальная миопатия - редкое генетическое заболевание
Содержание:
Митохондриальная миопатия – это одно из генетических заболеваний, которое в мире встречается относительно редко. Его особенность состоит в том, что причина болезни – это нарушение работы митохондрий, при этом степень тяжести заболевания будет зависеть от того, какое количество их не выполняет свою положенную работу. Существует несколько разновидностей этой болезни, и каждая из них имеет свои симптомы.
Причины
Как и любое другое заболевание, митохондриальная миопатия имеет свои причины. И здесь виноваты не вирусы или бактерии, и даже не действия беременной женщины или её окружения, а мутации в генах. А это значит, что такие гены с «поломками» ребёнок получает от своих родителей, а следовательно — эту болезнь можно назвать наследственной.
Хотя иногда бывает, что мутация в генах и рождение больного митохондриальной миопатией ребёнка происходит у совершенно здоровых родителей.
Когда клетка заполнена дефектными митохондриями, то она просто не в состоянии нормально работать. Кроме того, в ней могут постепенно накапливаться и не удаляться опасные вещества, которые в итоге приводят к смерти человека с этим диагнозом. А самыми чувствительными клетками к таким дефектам в митохондриях являются как раз мышечные и нервные. Именно они острее всего реагируют на полное отсутствие или нехватку такого вещества, как АТФ, а ведь именно его и должны вырабатывать митохондрии, для того, чтобы организм работал без перебоев.
Синдром Кирнса—Сейра
Это заболевание считается очень редким и является одним из вариантов митохондриальной миопатии. Первые признаки болезни приходятся на ранний детский или юношеский возраст, а среди основных симптомов можно отметить:
- Опущение век.
- Ограничение подвижности глазных яблок.
- Нарушения со стороны сердца.
- Атаксия.
- Тугоухость.
- Ретинопатия.
- Низкорослость.
- Деменция.
- Гипотиреоз.
- Сахарный диабет.
Эта патология обычно диагностируется до 20 лет. В некоторых случаях оно может прогрессировать очень быстро, иногда прогрессирование протекает медленно, но в итоге пациенты не доживают и до 40 лет. По наследству болезнь передаётся редко, чаще всего мутация в митохондриях происходит при внутриутробном развитии плода, по тем или иным причинам.
MERRF-синдром
Это заболевание стало известно не так давно. Среди его симптомов миоклонус-эпилепсия, миопатия, атаксия мозжечка, дизартрия, нистагм. Болезнь может начаться как у детей, так и у взрослых, развивается либо медленно, либо прогрессирует с большой скоростью. Человек, как правило, становится глубоким инвалидом до 40 лет.
МЕLАS-синдром
Это ещё одно состояние, которое входит в группу миопатий такого рода. Его трудно диагностировать, очень часто возникают ошибки в диагностике, поэтому сказать о распространённости недуга очень сложно.
Каждый случай имеет свои симптомы, так как к развитию этого синдрома приводит мутация в самых разных генах, из-за которой митохондрии перестают выполнять свои функции в полном объёме. Среди основных признаков выделяются миопатия, лактатацидоз, инсультоподобные эпизоды, проблемы с сердцем, судороги, отклонения в психике и непереносимость физической нагрузки.
Диагностика
Сложность диагностики этого недуга состоит в том, что для постановки правильного диагноза нужно провести сразу несколько анализов, результаты которых будут получены далеко не в течение одного дня.
Так, для диагностики любого из перечисленных синдромов обязательно проводится генеалогический анализ, то есть определяются родственники, у которых была или есть эта патология. Также делается клинический и биохимический анализ крови, анализ состояния мышц, для чего требуется биопсия, и, конечно — исследование генов, которое и даёт представление о том, где именно случилась «поломка», которая и привела к развитию такой миопатии.
Лечение
На сегодняшний день лечение митохондриальных миопатий не разработано. Но доказано, что в большинстве случаев пациентам продляет жизнь лечение при помощи витаминов. Например, известен случай, когда при помощи кофермента Q были сведены на нет признаки заболевания при МЕLАS-синдроме. Также в качестве лекарственных препаратов используются рибофлавин и никотинамид.
Чтобы предотвратить это наследственное заболевание, сегодня стали применять экстракорпоральное оплодотворение яйцеклеткой, ядро которой получено из яйцеклетки пациентки с присутствием митохондриальной миопатией, при этом цитоплазма в этой яйцеклетке использована от совершенно здоровой женщины, у которой митохондрии функционируют нормально. Однако такая замена пока находится в стадии разработки.
Митохондриальная миопатия | Центр лечения в Германии
Лечение митохондриальной миопатии в Германии.
Анамнез заболевания. Считает себя больной с пять лет. когда стала отмечать появившеюся шаткость при ходьбе, из-за чего не смогла бегать. После падения с потерей сознания состояние резко ухудшилось и постепенно нарастало до вышеописанных жалоб, психотравмирующей ситуации появилась снижение толерантности к физической нагрузке (длительная ходьба, поднимание по лестнице), стали беспокоить эпизодические головные боли с вовлечением перикраниальной мускулатуры, плохо купирующиеся анальгетиками.
Обследование пациента
Жалобы при поступлении на диагностику в Мюнхен: на шаткость при ходьбе из-за нарушения координации и слабости в дистальных отделах ног, сложности при спуске и подъеме по лестнице, похудание мышц предплечья и икроножных мышц, прогрессирующее ухудшение остроты зрения.
Соматический статус: состояние относительно удовлетворительное. Дыхание везикулярное, хрипов нет ЧД-16 в мин. Тоны сердца ясные, ритмичные ЧСС-84 уд. в мин., АД 110/70. Живот мягкий, безболезненный. Дизурии нет.
Неврологический статус: сознание ясное, контактна, ориентирована в месте, времени, личности. Менингиальных знаков нет. Движение глазных яблок в полном объеме. Зрачки D=S, реакция на свет живая. Глазные щели D=S. Корнеальные рефлексы сохранены. Лицо асимметрично из-за легкой сглаженности левой носогубной складки. Глотание, фонация не нарушены. Положение головы не нарушено. Язык по средней линии. Пассивные движения в полном объеме, активные движения ограничены в плечелопаточном суставе слева снижения мышечной силы в проксимальных отделах до 3.5-4 б. Гипотрофия плечелопаточных и височных мышц, межкостных мышц кистей с 2х сторон. Расстройств чувствительности нет. Сухожильные рефлексы снижены на руках, на ногах живые симметричны. Координаторных пробы выполняет удовлетворительно.
Результаты лабораторных анализов
Общий анализ крови: НЬ-13,9 г/дл, Ht-41,7% Эр- 4,78 х 10б/мл, лей- 7,4х 10 3 /мл, тромб-210х 103 /мл, п/я-1, с/я-45, баз-0, эоз-1, лим-51, мон- 2, СОЭ — 29 мм/ч.
Биохимический анализ крови: Г-ГТ-11 ед/л, АСТ-22 ед/л, АЛТ- 19 ед/л, общ. белок- 68,8 г/л, альбумин- 46,4 г/л, креатинин- 0.65 мг/дл, глюкоза- 4.3 ммоль/л, азот мочевины- 6.8 мг/дл, общ. билирубин 6.2 ммоль/л. белк. Фракции альбум- 58.3%, альфа-1- 4.1, альфа-2 — 9.7 %, бета- 10.4 %, гамма- 17.5 %, триглицериды- 0.75 ммоль/л, общ. холестерин- 5.72 ммоль/л, ЛПВП-хс 1.97 ммоль/л, ЛПНП-хс 3.6 ммоль/л, ЛПОНП-хс 0.14 ммоль/л, К А — 1,90. Тип гиперлипидемии 2а ВИЧ, гепатит В и С, сифилис не выявлены.
Результаты исследования биоптаты мышечной ткани: обнаружена мутация мтДНК А3243G, феномен «рваных красных волокон».
Проведена световая микроскопия гистохимическое выявления сукцинатдегидрогеназы NADH -тетразолий редуктазы, цитохромоксидазы.
Заключение по результатам диагностики митохондриальной миопатии в Германии: при световой микроскопии выявлены морфологические гистохимические признаки митохондриальной цитопатии.
Результаты инструментальной диагностики
ЭКГ: синусовый ритм, неполная блокада правой ветви п. Гиса, вариант нормы ЭКГ.
ЭНМГ, ЭМГ: исследованы срединный, малоберцовый нервы слева, большеберцовый и икроножный нервы справа, игольчатой ЭМГ исследованы m. Tibialis anterior sin.
Заключение. Проводимость по двигательным и чувствительным волокнам, параметры амплитуд моторных и сенсорных ответов исследованных нервов в пределах нормы. Признаков текущего денервационного процесса нет. Полученные результаты могут указывать ка на начальный мышечный уровень поражения, так и быть усугублены за счет общей гипотрофии мышц.
МРТ- головного мозга: картина расширения субарахноидальных пространств в лобных отделах.
Заключительный диагноз — митохондриальная миопатия.
На основании результатов лабораторных и инструментальных исследований предложена комплексная терапия по лечению митохондриальной миопатии в Германии.
На фоне проводимого лечения восстановилась координация, исчезла шаткость при ходьбе, головные боли перестали беспокоить, восстановился мышечный тонус. Была признана эффективность применяемого терапевтического курса лечения митохондриальной миопатии в Мюнхене и показано ее дальнейшее применение.
Рекомендовано
- Продолжить курс лечения митохондриальной миопатии.
- Наблюдение у невролога по месту жительства.
- ЛФК, ФТЛ курсами 2-3 раза в год, постоянное занятие в бассейне.
Миопатия: типы заболевания, причины, проявления
Миопатия - это общий термин для мышечного заболевания, которое не связано с каким-либо расстройством иннервации или нервно-мышечного соединения с широким спектром возможных этиологий.
Миопатия, Мышечная Дистрофия Излечима в АРМ!
Практически все виды миопатии приводят к ослаблению и атрофии скелетных мышц, особенно тех, которые находятся ближе всего к центру тела.
Миопатии можно разделить на наследственные и приобретенные. Наследственная группа включает:
- мышечные дистрофии,
- врожденные,
- метаболические,
- митохондриальные миопатии
- миотонии
- канелопатии.
И наоборот, воспалительные, эндокринные и токсические миопатии относятся к приобретенной группе.
Наследственные миопатии
Мышечные дистрофии представляют собой различную группу наследственных заболеваний, поражающих как детей, так и взрослых. Обусловлены 30-ю различными генами, ответственными за развитие заболевания. Патологии характеризуются истощением мышц и слабостью, с повышенным уровнем креатинкиназы (СК). Заболевания показывают дистрофический паттерн (то есть дегенеративный паттерн с некрозом и обширным фиброзом) и поражение центральной нервной системы.
Врожденные миопатии также являются генетически и клинически различной группой состояний, первоначально классифицированных в соответствии с уникальными морфологическими изменениями, наблюдаемыми в мышечной ткани. Тем не менее нет никаких некротических или дегенеративных изменений во врожденных миопатиях (в отличие от мышечной дистрофии), и уровни CK часто являются нормальными. Эта группа миопатий включает в себя некоторые хорошо известные состояния, такие как:
- нематическая миопатия,
- заболевание центрального ядра,
- миотубулярная миопатия с Х-хромосомой
- центронуклеарная миопатия.
Читайте также: Тортиколлис, или кривошея, у детей: причины появления
Метаболические миопатии представляют собой разнообразную группу расстройств, которые возникают в результате дефектов клеточного энергетического метаболизма, включая расщепление жизненно важных жирных кислот и углеводов с образованием аденозинтрифосфата. Следовательно, тремя основными категориями метаболических миопатий являются дефекты окисления жирных кислот, болезни накопления гликогена и нарушения митохондрий, связанные с нарушением дыхательной цепи.
Митохондриальные миопатии также представляют собой большую группу разнообразных расстройств, возникающих в результате первичной дисфункции дыхательной цепи митохондрий и впоследствии вызывающих мышечные заболевания. Эта группа болезней имеет множество различных фенотипов и генетических этиологий, и часто может иметь мультисистемную дисфункцию. Типичные примеры митохондриальных миопатий включают:
- тяжелый синдром Пирсона,
- синдром Кернса-Сэйра
- прогрессирующую внешнюю офтальмоплегию, которые могут проявляться в позднем взрослом возрасте.
Приобретенные миопатии
Идиопатические воспалительные миопатии представляют собой подмножество аутоиммунных заболеваний соединительной ткани, в первую очередь поражающих мышцы. Мышечная патология показывает характерные воспалительные экссудаты (жидкости, выделяющиеся в ткани или полости организма из мелких кровеносных сосудов при воспалении) переменного распределения внутри мышечного пучка, и есть различная степень повышения CK и раздражительной миопатии.
- По последним данным, с миопатией Дюшенна рождается один из 5000 мальчиков, а не один из 3000, как было принято считать раньше. Если переложить эту статистику на такой крупный город, как Москва, где за год рождаются около 100 тысяч детей, то каждый год должно рождаться 10 детей с миопатией Дюшенна. В среднем болезнь начинает проявляться в возрасте пяти лет. Дети испытывают значительные сложности при ходьбе, быстро устают, им сложно подниматься по лестнице, а при вставании с пола они применяют миопатические приемы Говерса (вставание «лесенкой»). Еще один важный симптом - большие голени. Члены семьи поначалу радуются, думая, что ребенок растет спортсменом, однако вскоре оказывается, что это не так, говорит и.о. руководителя Детского нервно-мышечного центра НИКИ педиатрии кандидат медицинских наук Дмитрий Влодавец.
Миопатия включена в число потенциальных побочных эффектов и токсичности, связанных с определенными липидоснижающими агентами (такими как ингибиторы 3-гидрокси-3-метилглутарил коэнзим А-редуктазы), но также с кортикостероидами или алкоголем. Тем не менее точный механизм статин-индуцированной мышечной токсичности остается неясным.
Фото: tahlil.com
Читайте также: Сильная головная боль у беременных: в чем опасность
причины, симптомы, диагностика и лечение
Метаболическая миопатия — целый комплекс заболеваний, обусловленных недостатком ферментов или нарушением обмена веществ в мышечной ткани. Для этих болезней свойственны такие симптомы, как мышечная слабость, судороги, боли, парезы и другие. Невролог обычно ставит диагноз на основании жалоб пациента, биопсии мышц, лабораторных анализов.
Содержание статьи:
Лечение патологий должно быть комплексным и включать в себя витаминотерапию, диетотерапию, ортопедическую коррекцию, прием медикаментов.
В группу метаболических миопатий включают множество врожденных и приобретенных болезней, которые встречаются как у детей, так и у взрослых. Статистические исследования доказывают, что метаболические миопатии развиваются в пяти случаях из тысячи. Прогноз для взрослых больных с приобретенной формой патологии более благоприятный, чем для пациентов с врожденной формой. Что касается детей, то для них в большинстве случаев прогноз заболевания неблагоприятен.
Причины развития метаболических миопатий
Метаболические миопатии могут иметь как врожденный, так и приобретенный характер. Что касается приобретенных заболеваний, то они в большинстве случаев бывают спровоцированы эндокринными патологиями: гипотиреоз, гипертиреоз, болезни Кушинга и Аддисона, гиперальдостеронизм, акромегалия. Причиной заболевания может также стать хроническая интоксикация организма из-за злоупотребления алкоголем или ятрогенного воздействия. В некоторых случаях метаболические миопатии возникают из-за хронических болезней печени (печеночная недостаточность, почек (хроническая почечная недостаточность, уремия, электролитные нарушения) и сердечно-сосудистой системы.
Главной причиной врожденных заболеваний в неврологии считают наследственную предрасположенность. Передача поврежденного гена в большинстве случаев осуществляется аутосомно-рецессивно, в редких случаях — по материнской линии. В практике неврологов были также зафиксированы спорадические и случайные генные мутации. Врожденные миопатии обусловлены патологиями ионных каналов мышечных мембран и нарушением нормального функционирования митохондрий.
Классификация метаболических миопатий
В неврологии выделяют первичные (врожденные) и вторичные (приобретенные) формы заболевания. Вторичные метаболические миопатии — последствие серьезных нарушений здоровья, в особенности эндокринных заболеваний. Первичные нарушения метаболизма мышечной ткани делят на несколько разновидностей в зависимости от причин, которые вызвали их появление.
В группу заболеваний, обусловленных нарушением обмена гликогена, входят такие патологии, как болезнь Андерсена, болезнь Помпе, болезнь Кори-Форбса, дефицит ФГК, болезнь Мак-Ардля, дефицит ЛДГ, болезнь Таури, дефицит фосфоглицеромутазы, дефицит киназы фосфорилазы b. Вызванные нарушением обмена липидов — недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина, дефицит КПТ. В случае нарушения метаболизма пуринов у больного диагностируют дефицит фермента МАДА. Выделяют также группу митохондриальных миопатий, в которую входят цитохрома b, b 1, дефицит ферментов редуктазы, дефицит АТФ.
Симптомы метаболической миопатии
Для клинической картины метаболической миопатии свойственны обширные симптомы. Тем не менее, их можно сгруппировать зависимо от того, какие нарушения в организме вызвало заболевание. В неврологии выделяют нарушения обмена глюкозы и гликогена, митохондриальные миопатии и нарушения липидного обмена.
Митохондриальные миопатии
Митохондриальные миопатии обычно диагностирую у детей и подростков. Патология поражает мышечные волокна глазного яблока и мышцы конечностей. В патологический процесс могут быть включены и другие органы: почки (тубулопатия), сердце (кардиомиопатии), печень, центральная нервная система (эпилептические припадки, деменции, мозжечковая атаксия, миоклонии), слуховой анализатор (нейросенсорная тугоухость). При физической нагрузке у больного могут наблюдаться одышка и головная боль, которая по своими проявлениям напоминает мигрень.
Для митохондриальной миопатии свойственна мышечная слабость, которая может иметь ограниченный и распространенный характер. В случае распространенного характера мышечной слабости пациент жалуется на плохую переносимость физических нагрузок и чрезмерную утомляемость. При ограниченном характере патологии наблюдается локальное поражение мышц лица, конечностей и глазного яблока.
Метаболическая миопатия
Метаболическая миопатия, как и большинство других неврологических патологий, сопровождается дефицитом липидных фракций. Зачастую это проявляется резким снижением способностей к выполнению физических упражнений. Наиболее распространенными случаями нарушения липидного обмена являются дефицит фермента КПТ и карнитина. Для синдрома дефицита карнитина свойственно прогрессирование мышечной слабости, поражение глоточных и лицевых мышц, а также мышц конечностей. При недостатке КПТ больной чувствует сильную боль из-за физических нагрузок.
Еще одним проявлением метаболической миопатии считается нарушение обмена глюкозы и гликогена. Если у пациента наблюдается избыточное скопление гликогена, то в состоянии покоя он чувствует себя нормально. В случае дефицита ферментов при повышении активности больные жалуются на мышечную слабость и чрезмерную утомляемость. Если в этом случае не прекратить физическое нагрузки, может произойти мышечная контрактура, что приведет к серьезному повреждению мышечной ткани.
Болезнь Мак-Ардля
Для болезни Мак-Ардля характерными будут сильные боли в мышцах, а также слабость и формирование уплотнений. Во время физической нагрузки у больных может случиться даже распад мышц, появиться признаки миоглобинурии и почечной недостаточности. Патология может также проявиться нарушением работы сердечно-сосудистой системы: сердечная недостаточность или слабость сердечной мускулатуры.
Болезнь Андерсена и Кори-Форбса
Имеет очень сложное течение, поскольку у пациента наблюдаются крайне тяжелые поражения печени и сердца. У больных детского возраста наблюдаются также умеренные поражения мышц и нарушения физического развития. Для младенческой формы заболевания характерен дефицит мальтазы. Обычно он развивается сразу после рождения малыша и сопровождается такими опасными для жизни симптомами, как застойные явления в сердце и мышечная слабость.
Практически всегда заболевание быстро прогрессирует и заканчивается смертью ребенка. Продолжительность жизни в случае дефицита мальтазы составляет не больше двух лет. Не менее тяжелыми проявлениями метаболической миопатии, которые также случаются в раннем возрасте, будет слабость нижних конечностей и серьезные дыхательные нарушения. Если дефицит мальтазы случается у взрослых пациентов, то у них наблюдается слабость нижних конечностей и мышечного пояса.
Диагностика метаболических миопатий
Постановка диагноза осуществляется на основании тщательного клинического обследования пациента, а также анамнеза заболевания. Наиболее выраженными клиническими признаками патологии считаются судороги, слабость, сильные мышечные боли, изменение оттенка мочи. Пациенту обязательно назначают дополнительно детальное неврологическое обследование. Для подтверждения диагноза невролог назначает исследование ферментативной активности: исследование крови на АСТ, КФК, АЛТ, ЛДГ.
Эффективными диагностическими методиками по праву считаются электронейрография и электромиография, которые позволяют точно определить наиболее характерные для миопатии отклонения. Для выявления различного рода органических изменений врачи назначают проведение КТ или МРТ мозга. По показаниям дополнительно может быть проведено томографическое обследование внутренних органов.
Наиболее точную информацию о заболевании позволяет получить биопсия мышц, а также последующее микроскопическое, морфологическое, гистохимическое исследование. Благодаря этой методике врачу удается поставить окончательный диагноз. Для постановки диагноза и дифференциальной диагностики неврологу может потребоваться консультация кардиолога, нефролога, офтальмолога, генетика, детского невролога. Крайне важно выявить заболевание на его ранней стадии, поскольку это позволяет подобрать оптимальный способ его лечения.
Лечение метаболических миопатий
На сегодняшний день не существует действенных методик лечения врожденных метаболических миопатий. Если у пациента была обнаружена такая форма заболевания, ему назначают только симптоматическую терапию. Больному необходимо будет соблюдать специальную диету, которая предусматривает преобладание в рационе продуктов с высоким содержанием белка. Показан также прием фруктозы и поливитаминов. Если у пациента была обнаружена миоглобинурия, ему дополнительно начинают вводить жидкость для предотвращения почечной недостаточности и поддержания нормального диуреза.
Для устранения симптомов митохондриальных миопатий пациенту приписывают медикаментозную терапию, предусматривающую прием препаратов, которые улучшают энергический метаболизм (убихинон, L-карнитин, тиамин, рибофлавин, витамины С и Е). По показаниям врача может быть также назначена ортопедическая коррекция. Лечение приобретенных заболеваний строится по иному принципу: терапия направлена не на устранение симптомов, а на лечение основного заболевания, вызвавшего метаболическую миопатию.
Прогноз и профилактика метаболических миопатий
Прогноз для пациентов с разными формами метаболических миопатий зачастую неблагоприятный. Особенно трудно поддается лечению заболевание, возникшее в первые недели жизни младенца. Младенческие формы метаболических миопатий имеют смертельный исход — обычно пациенты умирают в раннем возрасте. Что касается поздних форм заболевания, то они имеют куда более благоприятный прогноз. Положительный исход заболевания также зависит от вовлеченности в него патологий сердца, почек, печени и других органов.
Более благоприятный исход имеют приобретенные метаболические миопатии, поскольку после устранения факторов, которые их спровоцировали, можно полностью устранить все основные проявления болезни. Неврологи советуют будущим родителям соблюдать меры профилактики первичной (врожденной) патологии. Для этого перед зачатием малыша и в первом триместре беременности необходимо пройти медико-генетическое консультирование.
Миопатия. Формы, причины, симптомы и лечение миопатий.
Миопатия — прогрессирующая мышечная дистрофия — сборная группа заболеваний, характеризующихся первичным дистрофическим процессом в мышечной ткани.
Причины.
Относится к наиболее часто встречающимся хроническим заболеваниям нервно-мышечного аппарата и носит наследственный характер.
Различные экзогенные вредности (травмы, инфекция, интоксикация) могут выявить имеющуюся патологию или вызвать ухудшение текущего процесса. Для установления семейного характера заболевания необходим не только тщательный анамнез, но и по возможности более полный осмотр всех членов семьи с выявлением так называемых малых признаков заболевания.
Наличие спорадических случаев не исключает наследственную природу.
Следует также иметь в виду возможность фенокопий миопатии, т.е. симптоматических форм или миопатических синдромов.
Патологоанатомия.
При патологоанатомических исследованиях в нервной системе не находят характерных изменений. В редких случаях отмечаются незначительное уменьшение клеток передних рогов спинного мозга, иногда — изменения в двигательных нервных окончаниях в виде набухания миелиновой оболочки, изменения осевых цилиндров. В моторных бляшках исчезает фибриллярная структура. Грубые изменения находят в поперечнополосатых мышцах. Мышцы истончены, большая часть волокон замещена соединительной тканью и жиром. Характерна неравномерность отдельных мышечных волокон — одни волокна резко уменьшены, другие, наоборот, резко увеличены.
Нормальные, атрофированные и гипертрофированные волокна располагаются беспорядочно (в отличие от «пучковых» поражений при неврогенных амиотрофиях).
Как правило, отмечается увеличение числа мышечных ядер, которые формируются в цепочки. Гипертрофируются ядра сарколеммы.
Наблюдается продольное расщепление мышечных волокон и образование вакуолей. Уже в очень ранних стадиях заболевания обнаруживается значительное разрастание соединительной ткани, главным образом коллагеновых волокон.
В поздних стадиях заболевания почти вся мышечная ткань замещена соединительной или жировой тканью. В сосудах наблюдается пролиферация адвёнтиции, сужение просвета и иногда пристеночное тромбообразование. По мере развития процесса увеличивается общая масса эндо- и перимизиальной соединительной ткани с образованием фиброзного футляра вокруг мышечных волокон и внутримышечных кровеносных сосудов.
При гистохимическом исследовании наблюдается увеличение кислых муко- полисахаридов в основном веществе мышц и коллагеновых волокнах.
Патогенез.
Патогенез миопатий до настоящего времени неясен. Первичный биохимический дефект не установлен. Наиболее значительным изменениям подвергается белковый и углеводный обмен в мышечной ткани. В последнее время высказана гипотеза о нарушении обмена циклических нуклеотидов (циклических АМФ и ГМФ), являющихся универсальными регуляторами внутриклеточного обмена и ответственными за реализацию генетической информации.
Клиническая картина.
Симптоматика миопатий характеризуется нарастающими атрофиями произвольной мускулатуры. Параллельно развитию мышечного похудания появляются и парезы, однако мышечная слабость обычно выражена меньше, чем степень атрофии.
В связи с медленным прогрессированием процесса и неравномерностью поражения отдельных мышечных групп и даже участков мышц создаются условия для относительной компенсации двигательного дефекта: больные миопатией длительное время остаются трудоспособными и могут себя обслуживать, прибегая к ряду характерных вспомогательных движений. Постепенно угасают сухожильные рефлексы. Чувствительность, координация движений не нарушаются. Тазовые функции всегда сохранены.
Для некоторых видов миопатий характерны наличие псевдогипертрофий, наклонность к концевым атрофиям и сухожильным ретракциям. Фасцикулярные подергивания отсутствуют. Механическая возбудимость мышц снижена.
Нередко наблюдаются те или иные изменения внутренных органов,
- главным образом сердца: расширение границ, глухость тонов, нарушение проводимости, подтверждаемое электрокардиограммой;
- страдает функция внешнего дыхания;
- вегетативные нарушения выражаются цианозом кистей и стоп, резко повышенной потливостью, похолоданием дистальных отделов конечностей, асимметрией кожной температуры, пульса, повышенным пиломоторным рефлексом;
- страдает микроциркуляция в мышцах конечностей;
- рентгенография трубчатых костей позволяет обнаружить дистрофические явления;
- при электромиографическом исследовании констатируется характерная картина — снижение амплитуды биопотенциалов при достаточной частоте, а также укорочение длительности одиночного потенциала и полифазный характер;
- при биохимических исследованиях находят нарушения в креатин-креатининовом обмене. Почти всегда в моче значительно уменьшается количество креатинина и появляется креатин. Креатиновый показатель в известной степени говорит о тяжести дистрофического процесса. Отмечается значительная аминоацидурия. При ряде форм миопатий очень рано (в преклинической или в начальной клинической стадии) можно обнаружить увеличение ферментов в сыворотке крови. В первую очередь это касается специфического для мышечной ткани фермента креатинфосфокиназы;
- увеличивается также содержание аминофераз и альдолаз. Уменьшена артериовенозная разница содержания сахара крови, увеличено содержание пировиноградной и молочной кислот в мышцах и в крови. Как правило, уменьшен уровень лимонной кислоты в крови. Имеются данные о специфичности некоторых биохимических изменений при различных типах миопатий.
Классификация миопатий.
До настоящего времени нет достаточно обоснованной и общепринятой классификации миопатий. В большинстве случаев используют классификацию, основанную на клиническом принципе.
Псевдогипертрофическая форма Дюшенна является одной из наиболее частых форм миопатий. Она характеризуется самым ранним началом заболевания — нередко с 2—5-летнего возраста, а иногда даже с первого года жизни, и наиболее злокачественным течением. В типичных случаях дети к 10—12 годам уже с трудом ходят и к 15 годам становятся полностью обездвиженными.
В первую очередь страдают мышцы проксимальных отделов нижних конечностей, тазового пояса, затем в процесс вовлекаются мышцы проксимальных отделов рук. Рано выпадают коленные рефлексы. Характерна псевдогипертрофия икроножных мышц; часто уплотнение и гипертрофия их являются первым симптомом заболевания.
Псевдогипертрофии могут наблюдаться и в других группах мышц — ягодичных, дельтовидных, иногда - в языке; отмечаются значительно выраженные ретракции, в первую очередь со стороны ахилловых сухожилий. Нередко страдает сердечная мышца. Отмечается снижение интеллекта в разной степени выраженности. Для миопатии Дюшенна очень характерен высокий уровень ферментов сыворотки крови, особенно креатинфосфокиназы. Повышенный уровень ферментов можно обнаружить и у носительниц мутантного гена.
Заболевание передается по рецессивному, сцепленному с Х-хромосомой типу. Болеют только мальчики, матери являются кондукторами. Риск заболевания сыновей матерей-носительниц 50 %; 50 % дочерей становятся носителями патологического гена. Пенетрантность высокая.
Доброкачественная псевдогипертрофическая миопатия, сцепленная с Х-хромосомой (миопатия Беккера), выделена в самостоятельную форму в связи с рядом особенностей. Начало заболевания — чаще между 12 и 25 годами, иногда раньше. Течение мягкое, прогрессирование медленное, больные многие годы сохраняют трудоспособность или самообслуживание. Интеллект всегда сохранен. В остальном клиническая картина подобна псевдогипертрофической форме Дюшенна.
Плечелопаточно-лицевая форма Ландузи — Дежерина — относительно часто встречающийся тип миопатии.
Начинается заболевание, как правило, в детском или юношеском возрасте; течение сравнительно благоприятное. Первые симптомы касаются поражения мышц лица, особенно круговой мышцы рта, или мышц плечевого пояса. К этому присоединяется слабость и похудание мышц проксимальных отделов рук, затем развивается парез дистальных отделов ног. Наблюдаются умеренные гипертрофии мышц, а затем своеобразные патологические позы из-за неравномерности атрофии различных групп мышц и ретракций. Сухожильные рефлексы долго остаются сохраненными. Может быть асимметрия поражения.
Заболевание передается по аутосомно-доминантному типу с полной пенетрантностью, страдают в равной степени мужчины и женщины. Отмечаются отчетливые различия в тяжести клинических признаков не только в разных семьях, но и у разных членов одной семьи (могут быть тяжелые, легкие и абортивные формы).
Ювенильная форма Эрба, или мышечная дистрофия тазового и плечевого поясов — один из частых вариантов мышечной дистрофии. Характеризуется поражением, как правило, вначале мышц тазового пояса. Это проявляется изменением походки с раскачиванием («миопатическая походка»).
Как правило, страдают мышцы спины и живота (затруднение при вставании из положения лежа, сопровождаемое характерными вспомогательными движениями рук, усиленный лордоз, «лягушачий» живот). В дальнейшем поражаются мышцы плечевого пояса с развитием «крыловидных» лопаток. Отмечаются умеренные псевдогипертрофии, концевые атрофии, ретракция мышц.
Описаны стертые формы. В сыворотке крови находят умеренное повышение ферментов. Начало заболевания очень вариабельно— от детского возраста до сравнительно зрелого, но чаще в начале второго десятилетия, что отражает название этой формы. Также изменчив характер течения — иногда мягкое, благоприятное, иногда очень злокачественное.
Большинство авторов признают аутосомно-рецессивный тип наследования заболевания. Часты спорадические формы и фенокопии. Болеют одинаково часто мужчины и женщины.
Дистальная форма миопатии встречается редко.
Характеризуется поражением мышц голеней, стоп, предплечий, кистей, постепенно процесс генерализуется. Отмечаются ретракции, концевые атрофии мышц. Заболевание начинается в сравнительно позднем возрасте — 20—25 лет; прогрессирование, как правило, медленное. Отсутствие расстройств чувствительности, нормальная скорость проведения возбуждения, повышенный уровень сывороточных ферментов отличают дистальную миопатию от невральной амиотрофии.
Тип наследственной передачи—аутосомно-доминантный с неполной пенетрантностью. Несколько чаще болеют лица мужского пола.
Лопаточно-перонеальная амиотрофия (миопатия Давиденкова) проявляется поражением дистальных мышц нижних и проксимальных отделов верхних конечностей и мышц плечевого пояса. Начинается заболевание сравнительно поздно — в 25—30 лет. Наблюдаются концевые атрофии, например в большой грудной мышце, иногда крупные фасцикулярные подергивания, раннее угнетение сухожильных рефлексов.
В ряде случаев отмечаются легкие расстройства чувствительности — дистальные парестезии, гипестезии, иногда умеренная боль. Почти никогда не бывает креатинурии.
При электромиографическом исследовании выявляются специфические изменения, отличающие эту форму от обычной миопатии и невральной амиотрофии Шарко — Мари (дизритмичные колебания в покое, снижение амплитуды, уменьшение частоты и иногда групповые спайковые разряды при активных движениях).
Таким образом, данная форма является как бы промежуточной между первичной миопатией и невральной амиотрофией. Некоторые авторы рассматривают эту форму лишь как особый вариант (скапуло-перонеальный синдром) плечелопаточно-лицевой миопатии Ландузи — Дежерина.
Редкие варианты миопатии.
Описано большое количество различных вариантов прогрессирующего мышечного поражения наследственного характера. Так, например, описаны миопатия четырехглавой мышцы бедра, миосклеротическая миопатия, мышечная дистрофия с истинными гипертрофиями, врожденная мышечная дистрофия с медленным и быстрым прогрессированием, иногда с катарактой, мышечный инфантилизм и др.
Непрогрессирующие миопатии включают группу заболеваний, отличающихся или своеобразными изменениями строения мышечных клеток, или специфическими биохимическими нарушениями. Проявляются эти состояния сравнительно рано, обычно на 1—3-м году жизни, имеют сравнительно благоприятное течение. Диагноз может быть поставлен после биопсии мышц, иногда только после электронно- микроскопического исследования.
Болезнь центрального стержня характеризуется резким снижением или полным отсутствием ферментативной активности в центральной части мышечного волокна, что выявляется при окраске препарата мышечной ткани трехвалентным хромом по Гомори.
Клиническая картина: снижение мышечного тонуса, дряблость мышц, задержка развития двигательных функций, в позднем возрасте — умеренная слабость проксимальных отделов и гипотрофия мышц.
На ЭМГ — уменьшение длительности колебаний потенциала и увеличение полифазных потенциалов. Передача по доминантному типу с неполной пенетрантностью. Часты спорадические случаи.
Немалиновая, или нитеобразная, миопатия проявляется врожденной мышечной слабостью конечностей и лица с понижением мышечного тонуса и отсутствием сухожильных рефлексов. Описаны изменения скелета, в частности деформация грудной клетки, сколиоз.
На ЭМГ — изменения, характерные для мышечного уровня поражения. При электронно-микроскопическом исследовании выявляются своеобразные нитевидные структуры под сарколеммой.
Миотувулярная миопатия клинически выражается в понижении мышечного тонуса, умеренно выраженных атрофиях мышц конечностей с наличием диффузной слабости рук, ног, туловища. Характерны также слабость мимической мускулатуры, птоз и ограничение подвижности глазных яблок, общая задержка развития двигательных функций. Состояние может быть стационарным или медленно прогрессирующим. У большинства больных выявляются те или иные костные деформации.
На ЭМГ — сочетание мышечного типа изменений с наличием спонтанной активности. Гистологически определяются мышечные волокна резко уменьшенной величины с центральным расположением ядер, напоминающие по строению эмбриональную мышечную ткань. При электронной микроскопии выявляются участки дегенеративно измененных миофибрилл, при гистохимическом исследовании обнаруживается повышение активности митохондри- альных ферментов.
Митохондриальные миопатии характеризуются увеличением числа митохондрий в мышечных волокнах или увеличением размеров митохондрий, обнаруживаемых при электронно-микроскопическом исследовании. Клинически отмечается мышечная слабость, главным образом в проксимальных отделах рук и ног, вялость, быстрая утомляемость при отсутствии особых мышечных атрофий. Прогрессирования, как правило, не отмечается.
Диагноз прогрессирующей мышечной дистрофии, как правило, не представляет больших трудностей. Атипичные формы приходится дифференцировать от сирингомиелии (переднероговая форма), начальных явлений амиотрофического склероза, спинальных амиотрофий, полимиозита и других миопатических синдромов (см.). Комплексное обследование больного с применением биохимических (определение уровня ферментов и др.), электрофизиологических (ЭМГ, определение скорости распространения возбуждения по нерву), гистологических исследований и анализ клинической картины позволяют поставить правильный диагноз.
ЛЕЧЕНИЕ.
- Воздействие на энергетической обмен в мышцах осуществляется назначением АТФ в виде монокальциевой соли по 3—6 мл в день внутримышечно в течение 30 дней.
- Показано применение витамина Е внутрь по 30—40 капель 3 раза в день или внутримышечно раствор токоферола ацетата в масле по 1—2 мл — 20 инъекций (или эревит).
- Назначают витамины группы В, гликокол, лейцин (по 1 столовой ложке 3 раза в день), глутаминовую кислоту (по 0,5—1 г 3 раза в день).
Накопленный опыт по применению анаболических гормонов не подтверждает возлагавшихся на этот вид лечения надежд.
- Рекомендуется сочетать медикаментозное лечение с физиотерапией (гальванический воротник и гальванические трусы с кальцием, солянохвойные ванны со строго индивидуальной лечебной физкультурой при средней нагрузке).
- При наличии контрактур может быть рекомендовано оперативное вмешательство на сухожилиях.
- Показан систематически проводимый легкий массаж.