Анализ на полиморфизм генов что это такое
Полиморфизмы генов системы свертываемости крови
СОДЕРЖАНИЕ СТАТЬИ:
Полиморфизмы генов системы свертываемости крови не являются непосредственной и обязательной причиной развития заболевания, но могут обуславливать больший или меньший риск его развития при действии различных внешних факторов.
Поэтому при наличии полиморфизмов информируют о повышенном риске развития заболевания при гетерозиготном или гомозиготном носительстве полиморфизма. Риск развития заболевания измеряют отношением шансов OR (odds ratio).
Полиморфизмы генов системы свертываемости крови
В Европе официально проводят клиническое генетическое тестирование мутаций в генах: FV( Leiden), F2 (протромбин), PAI-1, MTHFR. Полиморфизмы генов системы свертываемости крови оказывают большое влияние на течение беременности, а если вы знаете о результатах этого анализа, вам будет проще запланировать вашу беременность.
Мутация Лейден 1691 G->A коагуляционного фактора V (F5)
Физиология и генетика
Коагуляционный фактор V или фактор V свертывания крови является белковым кофактором при образовании тромбина из протромбина. Полиморфизм G1691A Leiden (аминокислотная замена Arg (R) -> Gln (Q) в позиции 506, известная также как «мутация Лейден» или «Ляйден») является показателем риска развития венозных тромбозов.
Это точечная (однонуклеотидная) мутация гена, кодирующего фактор V свертывания крови, придает устойчивость активной форме фактора V к расщепляющему действию специализированного регулирующего фермента, С-белка, что приводит к гиперкоагуляции. Соответственно, риск образования тромбов повышается. Распространенность мутации в популяциях европейского типа составляет 2-6%.
Риск тромбозов глубоких вен (TГВ): в 7 раз выше у гетерозиготных носителей Лейденской мутации гена F5 Arg506Gln и в 80 раз выше у гомозигот.
Факторы, влияющие на развитие ТГВ
К первой группе факторов относится изменение гормонального статуса:
– Использование пероральных контрацептивов дополнительно повышает риск развития ТГВ в 30 раз у гетерозигот, в 100 раз при гомозиготном носительстве.
– Беременность – в 16 раз повышает риск ТГВ.
– Гормонзаместительная терапия – в 2-4 раза увеличивает риски.
Ко второй группе факторов относятся повреждения сосудов:
– Катетеризация центральных вен повышает риск ТГВ в 2-3 раза
– Хирургические вмешательства – в 13 раз.
К третьей группе факторов относится обездвиженность: постельный режим и длительные авиа-перелёты. Здесь лишь отмечается увеличение риска, но статистика должна быть более полной:
– Инфекционные и онкологические заболевания также повышают риск развития ТГВ. Риск развития ишемического инсульта у женщин в возрасте 18-49 лет при наличии Лейденской мутации возрастает в 2,6 раза, а на фоне приёма пероральных контрацептивов увеличивается в 11,2 раза.
Показания к анализу
- Венозный тромбоз,
- развитие тромбоэмболических заболеваний в молодом возрасте;
- рецидивирующий характер тромбоэмболизмов;
- сердечно-сосудистые заболевания в семейном анамнезе,
- заместительная гормонотерапия,
- прием гормональных контрацептивов,
- невынашивание беременности,
- фетоплацентарная недостаточность,
- внутриутробная гибель плода,
- токсикоз,
- задержка развития плода,
- отслойка плаценты,
- пациентам, готовящимся к большим полостным операциям (миома матки, кесарево сечение, кисты яичников и пр.).

Клинические данные
Наличие мутации Лейден повышает вероятность развития целого ряда осложнений беременности:
– выкидыш на ранних сроках (риск повышается в 3 раза),
– отставания развития плода,
– позднего токсикоза (гестоза),
– фетоплацентарной недостаточности.
Повышенная склонность к тромбообразованию может приводить к артериальным тромбоэмболиям, инфаркту миокарда и инсульту. Наличие мутации Лейден повышает риск первичных и рецидивирующих венозных тромбозов, по крайней мере, в 3-6 раз.
Приводимые ниже примеры иллюстрируют связь мутации с различными видами тромбозов и другими кардиоваскулярными заболеваниями.
В течение 8 лет в нескольких центрах проводилось исследование более 300 пациентов с венозной тромбоэмболией (ВТЭ), в ходе которого был установлен повышенный 3,7-кратный риск ВТЭ при наличии мутации Лейден. В другой работе пациенты с венозной тромбоэмболией исследовались в течение 68 месяцев. За это время 14% пациентов перенесли рецидив ВТЭ.
Мутация Лейден фактора V приводит к четырехкратному увеличению риска повторного ВТЭ. Для пациентов с ВТЭ, имеющим мутацию Лейден, рекомендована более длительная антикоагуляционная терапия по сравнению с пациентами с нормальным фактором V.
Следует отметить, что риск развития венозных тромбозов значительно увеличивается (8-кратное увеличение), если пациент, кроме мутации Лейден фактора V, также имеет мутацию Т полиморфизма С677Т гена метилтетрагидрофолатредуктазы.
Одним из самых опасных осложнений гормональных контрацептивов являются тромбозы и тромбоэмболии. Многие женщины с такими осложнениями являются гетерозиготными носителями мутации Лейден (генотип G/A). На фоне приема гормональных контрацептивов риск тромбозов у них повышен в 6-9 раз.
У женщин, использующих гормональные противозачаточные средства и имеющих гомозиготную мутацию Лейден (генотип A/A), риск развития тромбоза церебральных синусов (ТЦС) повышен более чем в 30 раз по сравнению с пациентками, не имеющих этой мутации.
Были обобщены конечные данные исследования «Women’s Health Initiative Estrogen Plus Progestin» о частоте венозных тромбозов на фоне заместительной гормонотерапии (ЗГТ). В исследовании приняли участие 16 608 женщин в постменопаузе в возрасте от 50 до 79 лет, наблюдавшихся с 1993 по 1998 гг. в течение 5 лет. Наличие мутации Лейден усиливало риск тромбозов при эстроген-гестагенной заместительной гормонотерапии почти в 7 раз по сравнению с женщинами без этой мутации.
Присутствие других генетических мутаций (протромбин 20210А, метилентетрагидрофолатредуктаза С677Т, фактор XIII Val34Leu, PAI-1 4G/5G, фактор V HR2) не влияли на связь ЗГТ и риска венозных тромбозов. Анализ более десяти независимых исследований показал, что среди пациентов, перенесших инфаркт миокарда до 55 лет, распространенность мутации Лейден была заметно выше.
Среднестатистический риск развития инфаркта миокарда увеличивается в 1,5 раза. Более того, мутация Лейден приводит к 2,8 – кратному повышению количества пациентов без выраженного коронарного стеноза, заболевших инфарктом миокарда.
Полиморфизм 20210 G->A протромбина
Физиология и генетика
Протромбин (коагуляционный фактор II или F2) является одним из главных компонентов системы свертываемости крови. В ходе ферментативного расщепления протромбина образуется тромбин. Данная реакция является первой стадией образования кровяных сгустков.
Мутация гена протромбина G20210A характеризуется заменой нуклеотида гуанина (G) нуклеотидом аденин (A) в позиции 20210. Из-за увеличения экспрессии мутантного гена уровень протромбина может быть в полтора-два раза выше, чем в норме. Мутация наследуется по аутосомно-доминантному типу. Это означает, что тромбофилия возникает даже у гетерозиготного носителя измененного гена (G/A).
Тромбоэмболические заболевания (ТЭ) вызываются нарушениями в системе свертываемости крови. Эти нарушения приводят и к сердечно-сосудистым заболеваниям. Генотип G/A является показателем риска развития тромбозов и инфаркта миокарда. При возникновении тромбозов мутация 20210A часто встречается в сочетании с мутацией Лейден.
Генотип G/A позиции 20210 гена протромбина является фактором риска тех же осложнений, которые связанны с мутацией Лейден.
Гетерозиготными носителями гена являются 2-3% представителей европейской расы.
Риск развития ТГВ у носителей мутантного аллеля (А) гена F2 повышен в 2,8 раз. Комбинация мутации протромбина с Лейденской мутацией дополнительно увеличивает риски.
Согласно рекомендациям для акушеров и гинекологов (Великобритания, 2000г.), клинический генетический анализ FV и протромбина 20210 уместен из-за разных рисков гомозигот и гетерозигот.
Различают очень высокую, высокую и среднюю степень риска венозных тромбозов у беременных:
– Высокая степень риска у женщин с индивидуальной и семейной историей тромбозов и гомозиготных по Лейденской мутации, мутации G20210A протромбина или комбинацией этих мутаций. Таким пациенткам показана антикоагуляционная терапия низкомолекулярными гепаринами с начала-середины второго триместра.
– Средняя степень риска у женщин с семейной историей тромбозов и гетерозиготных по Лейденской мутации или мутации G20210A.В этом случае антикоагуляционная терапия не показана.
Показания к анализу
- Инфаркт миокарда,
- повышенный уровень протромбина крови,
- тромбоэмболические заболевания в анамнезе,
- преклонный возраст пациента,
- невынашивание беременности,
- фетоплацентарная недостаточность,
- внутриутробная гибель плода,
- токсикоз,
- задержка развития плода,
- отслойка плаценты,
- пациентам, готовящимся к большим полостным операциям (миома матки, кесарево сечение, кисты яичников и пр.), курение.
Клинические данные
В исследовании 500 пациентов с инфарктом миокарда и 500 здоровых доноров показано более чем пятикратное увеличение риска инфаркта миокарда у пациентов с генотипом 20210A моложе 51 года. Генетический анализ группы пациенток с первым инфарктом миокарда (возраст 18-44 лет) показал, что вариант 20210А встречается в четыре раза чаще в сравнении с группой здоровых, что соответствует увеличению риска инфаркта в 4 раза.
Вероятность инфаркта была особенно высока при наличии других факторов риска сердечно-сосудистых заболеваний. Например, курение при наличии генотипа 20210А повышает риск инфаркта миокарда более чем в 40 раз. Мутация 20210А является значительным фактором риска раннего инфаркта миокарда.
При исследовании пациентов с семейным анамнезом венозного тромбоза и контрольной группы здоровых доноров обнаружено, что мутация 20210A приводит к трехкратному увеличению риска венозного тромбоза. Риск тромбоза увеличивается для всех возрастов и для обоих полов. В этом исследовании также была подтверждена прямая связь между наличием мутации 20210A и повышенным уровнем протромбина в крови.
В терапевтических стационарах, где преобладают больные с сердечно-сосудистыми заболеваниями, ТЭ в форме тромбоэмболии легочной артерии встречается в 15-30% случаев. Во многих случаях ТЭ являются непосредственной причиной смерти, особенно у послеоперационных больных и больных раком.
Установлено, что среди больных раком при наличии ТЭ смертность увеличивается в несколько раз, при этом количество ТЭ превышает среднестатистические значения. Причины роста ТЭ у больных раком, возможно, следует искать в проводимой терапии, несогласованной с генетической предрасположенностью больного. Это касается не только больных раком. Согласно патологоанатомическим отчетам, у 60% больных, умерших в больницах общего профиля, обнаруживают признаки тромбоэмболических заболеваний.
Знание генотипических характеристик пациента позволит не только оценить риск развития угрожающих жизни состояний, но и правильно определить способы их профилактики и лечения, а также возможность применения тех или иных лекарственных препаратов.
Термолабильный вариант A222V (677 С->Т) метилентетрагидрофолатредуктазы
Физиология и генетика
Метилентетрагидрофолатредуктаза (MTHFR) играет ключевую роль в метаболизме фолиевой кислоты. Фермент катализирует восстановление 5,10-метилентетрагидрофолата в 5-метилтетрагидрофолат.
Последний является активной формой фолиевой кислоты необходимой для образования метионина из гомоцистеина и далее — S-аденозилметионина, играющего ключевую роль в процессе метилирования ДНК. Дефицит MTHFR способствует не только тератогенному (повреждающему плод), но и мутагенному (повреждающему ДНК) действию.
При этом происходит инактивация многих клеточных генов, в том числе — онкогенов. В этом заключается одна из причин, по которой онкологи заинтересовались генетическими вариантами MTHFR. Аминокислота гомоцистеин является промежуточным продуктом процесса синтеза метионина. Нарушения фермента MTHFR приводят к избыточному накоплению гомоцистеина в плазме крови — гипергомоцистеинемии.
Ген MTHFR локализован на хромосоме 1р36.3. Известно около двух десятков мутаций этого гена, нарушающих функцию фермента. Наиболее изученной мутацией является вариант, в котором нуклеотид цитозин (C) в позиции 677 заменен тимидином (T), что приводит к замене аминокислотного остатка аланина на остаток валина (позиция 222) в сайте связывания фолата.
Такой полиморфизм MTHR обозначается как мутация C677T. У лиц, гомозиготных по данной мутации (генотип Т/Т), отмечается термолабильность MTHFR и снижение активности фермента примерно до 35% от среднего значения. В целом по населению земного шара, мутация 677Т гена MTHFR распространена достаточно широко у представителей европейской (кавказской) расы.
Были изучены частоты двух основных мутаций (С677Т и А1298С) среди представителей населения США. Показано наличие гомозиготы Т/Т у 10-16% европейцев и 10% лиц испанского происхождения, а гетерозиготными носителями этого гена были, соответственно, 56 и 52% обследованных лиц, т.е. наличие варианта 677Т (генотипы С/Т или Т/Т) наблюдалось в 62-72% случаев.
Аналогичные результаты были получены в отношении европейских выборок населения. Полиморфизм C677T связан, по крайней мере, с четырьмя группами многофакторных заболеваний: сердечно-сосудистыми заболеваниями, дефектами развития плода, колоректальной аденомой и раком молочной железы и яичника.
Показания к анализу
- Повышенный уровень гомоцистеина крови (гипергомоцистеинемия),
- сердечно-сосудистые заболевания (в частности, ишемическая болезнь сердца (ИБС) и инфаркт миокарда),
- атеросклероз,
- атеротромбоз
- Антифосфолипидный синдром
- Химиотерапия рака до или в процессе беременности
- Семейная предрасположенность к осложнениям беременности, приводящим к врожденным порокам развития: дефектам нервной системы плода, анэнцефалии, деформации лицевого скелета (волчья пасть, заячья губа), пренатальная смерть плода
- Полипоз кишечника, колоректальная аденома при употреблении алкоголя, рак прямой кишки
- Семейная предрасположенность к онкологическим заболеваниям, наличие мутаций генов BRCA
- Цервикальная дисплазия, особенно в сочетании с папилловирусными инфекциями.
Клинические данные
Дефекты в данном гене часто приводят к различным заболеваниям с широким спектром клинических симптомов: умственное и физическое отставание в развитии, пренатальная смерть или дефект плода, кардиоваскулярные и нейродегенеративные заболевания, диабет, рак и другие.
У носителей гетерозигот С/Т во время беременности наблюдается дефицит фолиевой кислоты, что может приводить к дефектам развития нервной трубки у плода. Курение усиливает влияние мутации. У носителей двух аллелей Т/Т (гомозиготное состояние) особенно высок риск развития побочных эффектов при приеме лекарственных препаратов, используемых в химиотерапии рака.
Гипергомоцистеинемия (ГГ) является независимым фактором риска атеросклероза и атеротромбоза (независимым от гиперлипидемии, гипертензии, сахарного диабета и т.д.). Установлено, что 10% риска развития коронарного атеросклероза обусловлено повышением уровня гомоцистеина в плазме крови. При исследовании группы пациентов с ГГ и группы здоровых доноров гомозиготная форма 677T была найдена у 73% пациентов с ГГ и только у 10% здоровых доноров.
Наличие гомозиготной формы 677T приводит к почти 10-кратному повышению риска ГГ. Пациенты с ГГ также имели пониженные уровни фолиевой кислоты и витамина В12, потребляли больше кофе и курили чаще, чем здоровые доноры. В норме уровень гомоцистеина равен 5-15 мкмоль/л, умеренно-повышенный уровень 15-30 мкмоль/л.
При тяжелой форме ГГ возможно 40-кратное повышение уровня гомоцистеина. Исследователи приписывают причину возникновения тяжелых форм ГГ и другим мутациям и факторам – гомозиготная мутация гена Cb S, самыми частыми считают I278T и G307S, хотя частота их проявления в разных странах сильно варьирует, намного реже причинами тяжелой ГГ являются Т/Т генотип MTHFR, дефицит метионинсинтетазы и нарушенная активность метионинсинтетазы из-за генетических нарушений метаболизма витамина В12.
Коррекцию ГГ можно провести поступлением кофакторов, необходимых для метаболизма гомоцистеина (фолиевая кислота, витамины В12, В1 и В6 (особенности терапии ГГ витаминами). У носителей Т/Т генотипа MTHFR при оптимальном потреблении фолата уровень гомоцистеина повышен умеренно (до 50%).
Хотя известно, что при тяжелой форме ГГ комбинация 2,5 мг фолиевой кислоты, 25 мг витамина В6 и 250 мкг витамина В12 в день снижает прогрессирование атеросклероза (измерялась бляшка в сонной артерии), нужно еще подтвердить, предупреждает ли гомоцистеин-снижающая терапия значимые сосудистые осложнения у лиц с умеренной ГГ.
О важности проблемы ГГ говорит тот факт, что Министерство здравоохранения США в 1992 году рекомендовало женщинам, которые могут забеременеть, принимать 400 мкг фолиевой кислоты в день.
Администрация по контролю пищевых продуктов и лекарственных препаратов в США требует обогащать крупы фолиевой кислотой в концентрациях, которые могут дать дополнительно 100 мкг в день. Однако, дневная доза фолиевой кислоты, необходимая для максимального снижения уровня гомоцистеина, равна 400 мкг, то есть могут быть оправданы и более высокие дозы добавок фолиевой кислоты в пищу.
Патогенез врожденных дефектов нервной трубки включает в себя, в частности, генетические и диетические факторы. При исследовании 40 детей Южной Италии с врожденным дефектом нервной трубки и здоровых доноров было показано, что генотип 677С в гомозиготном состоянии (С/С) приводит к двукратному повышению риска развития дефектов, в то время как мутантная гомозигота Т/Т соответствует почти десятикратному снижению риска.
При исследовании выборки населения Ирландии (395 больных и 848 здоровых) было установлено, что встречаемость варианта T повышена у пациентов с врожденным дефектом нервной трубки. Трудно сказать связаны ли эти противоположные результаты исследований с популяционными изменениями или не учтены другие факторы риска. Поэтому пока нельзя определить является ли вариант Т защитным или, наоборот, патогенным фактором для данного заболевания.
Повышение частоты генотипа 677T было отмечено не только при позднем токсикозе (гестозе), но и при других осложнениях беременности (отслойке плаценты, задержке роста плода, пренатальной смерти плода). Сочетание мутации 677T с другими факторами риска приводит к повышению вероятности раннего выкидыша.
При исследовании связи между мутацией 677T и сердечно-сосудистыми заболеваниями обнаружено, что гомозиготная мутация 677T встречается гораздо чаще у пациентов с кардиоваскулярными заболеваниями, чем у здоровых доноров. У молодых пациентов, имевших ишемию артерий, гомозигота Т/Т встречается в 1,2 раза чаще.
Статистический анализ 40 независимых исследований (мета-анализ) пациентов с ИБС, обобщающий данные о 11162 пациентах и 12758 здоровых доноров, показал увеличение риска развития ИБС в 1,16 раза при наличии гомозиготы Т/Т. Невысокая степень риска связана с гетерогенностью анализируемых выборок населения.
При исследовании гомогенных выборок населения (индивидуальные исследования, а не мета-анализ) оценки степени риска значительно выше. Так, разница в частотах встречаемости гомозигот Т/Т у пациентов и у здоровых доноров соответствовала 3-х кратному повышению риска кардиоваскулярных заболеваний в раннем возрасте. Наличие мутации 677Т в гене MTHFR у больных с антифосфолипидным синдромом коррелирует с рецидивирующим течением тромбозов.
Выявлена определенная, хотя и сложная, взаимосвязь между вариантами MTHFR и развитием предраковых и раковых состояний колоректальной области. Проводилось исследование значительной группы больных с полипозом толстого кишечника. Определялись уровни фолата в эритроцитах, наряду с оценкой С/Т генотипа МТHFR. Ранее полученные результаты показывали связь между пониженным содержанием фолата и риском развития аденоматоза.
Многофакторный анализ показал, что курение, фолатный статус и генотип MTHFR являются существенными компонентами высокого риска аденоматоза. Этот риск оказался весьма велик у лиц с низким уровнем фолата и носительством аллеля 677Т в гомо- или гетерозиготной форме. Эти данные показали сильное взаимодействие диетических и генных факторов в развитии предраковых состояний.
Сходные предположения выдвинуты учеными, которые обследовали большой контингент больных раком толстого кишечника и показали значительную связь между риском развития ракового заболевания, возрастом больных, возрастным дефицитом фолата и Т/Т генотипом MTHFR.
Исследование 379 пациентов с колоректальной аденомой и 726 здоровых доноров показало, что мужчины-носители Т/T генотипа, употребляющие много алкоголя, имели в 3,5 раза более высокий риск заболевания аденомой. Однако некоторые исследователи считают, что без употребления алкоголя, как одного из факторов риска, мутация 677T является защитным фактором.
Так, исследование пациентов с проксимальным колоректальным раком показало, что наличие у пациента гомозиготы Т/T приводит к 2,8-кратному понижению риска развития колоректального рака. Эти выводы требуют проверки для других популяций.
Скорее всего, значимость малоактивного мутантного MTHFR можно считать усугубляющей на фоне остальных перечисленных факторов риска, поскольку этот генный дефект может снижать стабильность генома из-за гипометилирования ДНК. Полиморфизм С677T влияет на эффективность химиотерапии рака. Фторурацил широко используется для химиотерапии колоректального рака.
Вероятность положительной динамики в ответ на химиотерапию колоректальной аденокарциномы при наличии у пациента 677T генотипа увеличивалась почти в три раза. Результаты позволяют предположить, что генотипирование по полиморфизму С677T позволит разработать более эффективные курсы химиотерапии.
Однако исследование небольших выборок (до 50) больных раком груди показало, что при наличии гомозиготы Т/Т риск развития побочных эффектов при применении метотрексата (антиметаболита, действие которого связано с ингибированием активности фермента MTHFR) увеличивается в десятки раз.
Имеются немногочисленные исследования генотипа MTHFR при онкогинекологических заболеваниях. Изучался полиморфизм С677Т гена MTHFR в большой группе еврейских женщин, заболевших раком молочной железы и яичника, включая и наследственные формы, связанные с мутациями генов BRCA. При таком неблагоприятном генетическом фоне наличие у больных Т/Т генотипа оказалось существенным фактором отягощения заболевания.
Частота Т/Т генотипа была в 2 раза выше (33% против 17%, Р=0,0026) среди женщин с двусторонним раком молочной железы и раком яичника, по сравнению с основной группой больных. Женщины с гетерозиготным генотипом С/Т имели двойной онкологический риск, а у больных с гомозиготным генотипом Т/Т риск был повышен втрое по сравнению с контрольной группой.
В то же время, пониженное потребление фолатов в диете повышало генетический риск до пятикратного значения по сравнению с контролем. Авторы также подтвердили тот факт, что заражение HPV (вирус папилломы) у больных является важнейшим фактором риска развития цервикальной дисплазии. В то же время подчеркивается особое значение сочетания HPV-инфекции с Т/Т вариантом MTHFR.
Полиморфизм Arg353Gln (10976 G->A) коагуляционного фактора VII (F7)
Физиология и генетика
В активном состоянии фактор VII взаимодействует с фактором III, что приводит к активации факторов IX и X системы свертывания крови, то есть коагуляционный фактор VII участвует в образовании кровяного сгустка.
Вариант 353Gln (10976A) приводит к понижению производительности (экспрессии) гена фактора VII и является защитным фактором в развитии тромбозов и инфаркта миокарда. Распространенность данного варианта в европейских популяциях составляет 10-20%.
Показания к анализу
- Риск инфаркта миокарда и фатального исхода при инфаркте миокарда,
- уровень коагуляционного фактора VII в крови,
- тромбоэмболические заболевания в анамнезе.

Клинические данные
Высокий уровень коагуляционного фактора VII в крови связывают с повышенным риском смерти при инфаркте миокарда [Meade TW et al, Lancet 1986, 2: 533-7].
Приведенные данные о клинической значимости мутации подтверждаются исследованиями в других европейских популяциях. В частности, наличие варианта 10976A соответствовало пониженному риску фатального исхода при инфаркте миокарда.
При исследовании пациентов со стенозом коронарных артерий и инфарктом миокарда обнаружено, что наличие мутации 10976A приводит к понижению уровня фактора VII в крови на 30% и 2-х кратному понижению риска инфаркта миокарда даже при наличии заметного коронарного атеросклероза.
В группе пациентов, не имевших инфаркта миокарда, наблюдалась повышенная встречаемость гетеро- и гомозиготных генотипов 10976A, соответственно G/A и G/G.
Полиморфизм – -455 G->A фибриногена
Физиология и генетика
При повреждении кровеносных сосудов фибриноген переходит в фибрин — основной компонент кровяных сгустков (тромбов). Мутация -455А бета фибриногена (FGB) сопровождается повышенной производительностью (экспрессией) гена, что приводит к повышенному уровню фибриногена в крови и увеличивает вероятность образования тромбов. Распространенность данного варианта в европейских популяциях составляет 5-10%.
Показания к анализу
- Повышенный уровень фибриногена плазмы крови,
- повышенное давление крови,
- тромбоэмболические заболевания в анамнезе,
- инсульт
Клинические данные
Повышенная склонность к тромбообразованию может приводить к тромбозам и кардиоваскулярным заболеваниям. Уровень фибриногена в крови определяется рядом факторов, среди которых прием лекарственных препаратов, курение, прием алкоголя и вес тела. Однако и генотипам G и A соответствует заметная разница в уровнях фибриногена крови (10-30% по различным исследованиям).
В исследовании группы здоровых доноров было установлено, что мутация -455А приводит к повышенному содержанию фибриногена в крови. В крупномасштабном исследовании EUROSTROKE было установлено, что риск инсульта (ишемического или геморрагического) повышается в 2-3 раза при увеличении содержания фибриногена крови. Риск дополнительно увеличивается при повышенном систолическом давлении (>160 мм рт. ст.). Эти данные подтверждаются исследованиями неевропейских популяций.
При повышенном давлении крови наличие генотипа -455А повышает риск развития ишемического инсульта.
Пациенты с инсультом, имеющие генотип -455А, характеризуются многоочаговостью поражений: могут иметь три или более лакунарных инфаркта церебральных сосудов, в среднем риск инсульта увеличивается в 2,6 раза.
При повышенном давлении крови у пациентов с мутацией риск многоочагового инсульта повышается более чем в 4 раза ([12637691], Финляндия).
Полиморфизм – IIeMet (66 a-g) Мутация редуктазы метионинсинтетазы
Физиология и генетика
Ген MTRR кодирует фермент метионин-синтазу редуктазу (МСР), участвующий в большом количестве биохимических реакций, связанных с переносом метильной группы. Одной из функций МСР является обратное превращение гомоцистеина в метионин. В качестве кофактора в этой реакции принимает участие витамин В12 (кобаламин).
Полиморфизм I22M A->G связан с аминокислотной заменой в молекуле фермента МСР. В результате этой замены функциональная активность фермента снижается, что приводит к повышению риска нарушений развития плода – дефектов невральной трубки. Влияние полиморфизма усугубляется дефицитом витамина В12. При сочетании полиморфизма I22M A->G гена MTRR с полиморфизмом 677C-> T в гене MTHFR риск увеличивается.
Полиморфизм I22M A->G гена MTRR также усиливает гипергомоцистеинемию, вызываемую полиморфизмом 677C-> T в гене MTHFR. Полиморфизм A66G (Ile22Met) в гене MTRR как в гетерозиготном (AG), так и в гомозиготном (GG) вариантах значительно повышает концентрацию гомоцистеина только при одновременном сочетании с генотипом MTHFR 677TT.
Полиморфизм MTRR 66 A-G увеличивает риск рождения ребенка с синдромом Дауна в 2,57 раз. Сочетание полиморфизмов в генах MTHFR и MTRR повышает этот риск до 4,08%.
Полиморфизм – 675 5G/4G Мутация ингибитора активатора плазминогена (PAI) 1
Физиология и генетика
Этот белок (известный также как SERPINE1 и PAI-1) один из основных компонентов тромболитической плазминоген-плазминовой системы. PAI-1 ингибирует тканевой и урокиназный активаторы плазминогена. Соответственно, PAI-1 играет важную роль в предопределении расположенности к кардиоваскулярным заболеваниям.
Гомозиготный вариант 4G полиморфизма –675 4G/5G является фактором риска развития тромбозов и инфаркта миокарда. Распространенность гомозиготной формы данного варианта в европеиоидных популяциях составляет 5-8%. Ген PAI-1 отличается от всех известных генов человека максимальной реакцией на стрессовые воздействия. Связь мутантного аллеля 4G с повышенным риском ТГВ анализировали во многих исследованиях, но их результаты носят противоречивый характер.
По данным российских исследователей (Ст.-Петербург) риск развития церебральных тромбозов возрастал у лиц с семейной историей сердечно-сосудистых заболеваний при наличии 4G аллеля в 6 раз. Показана ассоциация носительства полиморфизма 4G с привычным невынашиванием беременности.
Клинические аспекты
Вариант 4G приводит к повышенной экспрессии гена и, следовательно, к повышенному уровню PAI-1 в крови. Следовательно, тромболитическая система заторможена и риск тромбообразования возрастает.
В исследовании больших выборок населения (357 пациентов и 281 здоровых доноров) было установлено что вариант 4G/4G повышает риск развития тромбозов в среднем в 1.7 раза. Повышение риска было гораздо выше для подгрупп пациентов с тромбозом портальной вены и тромбозом внутренних органов.
Однако, не было установлено статистически значимых корреляций для подгрупп пациентов с глубоким тромбозом вен, церебральным или ретинальным тромбозами. Вариант 4G был ассоциирован с повышенным риском инфаркта миокарда. При наличии варианта 4G в PAI-1 и L33P в гене ITGB3 среднестатистический риска развития инфаркта миокарда повышался в 4.5-раза, у мужчин риск повышался в 6 раз при наличии этих двух вариантов.
Исследование 1179 здоровых доноров и их близких родственников показало вариант 4G ассоциирован с наличием семейной истории коронарной болезни артерий и/или сердца. В этом исследовании большой выборки население среднестатистическое повышение риска при наличии гомозигот составило 1.6 раза. Варианты полиморфизма 4G/5G особенно заметно коррелируют со средними уровнями PAI-1 в крови при наличии ожирения.
Было высказано предположение что влияние варианта 4G связано скорее с центральным а не с периферальным ожирением. Так как пациенты с центральным ожирением в особенности подвержены риску кардиоваскулярных заболеваний, влияние полиморфизма на уровень PAI-1 крови может приводить к дополнительному увеличению риска.
Показания к анализу полиморфизма
- Тромбоз портальной вены,
- тромбоз внутренних органов,
- инфаркт миокарда,
- семейная история инфаркта миокарда,
- коронарная болезнь артерий/сердца,
- уровень PAI-1 крови,
- ожирение.
Генетические анализы при потерях беременности
Aнализы на генетические полиморфизмы при выкидышах и замерших беременностях в первом триместре
Поскольку 80%[1] потерь приходятся на первые три месяца беременности, только этих ситуаций мы коснемся в статье. Причины потери беременности после 12 недель – предмет отдельного обсуждения.

Непросто поверить, но риск спонтанного прерывания беременности на сроке 6-12 недель у здоровой женщины моложе 35 лет составляет не менее 10%[2], и повлиять на причины этих событий можно в меньшинстве случаев.
Итак, женщина понимает: ребенка не будет. Одно из первых желаний в такие моменты – узнать причину. И находятся желающие эту потребность удовлетворить: ошарашенной женщине назначают многочисленные обследования и анализы, и редко обходится без тестов на:
- «генетические причины потери беременности»
- «полиморфизмы в генах гемостаза и фолатного цикла»
- «мутации предрасположенности к невынашиванию беременности»
- «генетический риск осложнений беременности»
- …
Вариантов названий у этой услуги множество. Суть одна: по материалу матери определяют ее генотип по полиморфизмам нескольких генов.
Когда женщине назначают «генетические анализы» по поводу потери беременности — это в 99% случаев именно анализы на полиморфизмы. Поэтому (с определенным допущением) можно сказать, что анализы на полиморфизмы = генетические анализы, которые назначаются женщинам по поводу потери беременности.
| Непросто в двух словах рассказать, что такое полиморфизмы. Полиморфизмы – это незначительные различия в структуре генов, определяющие разнообразие их проявлений. Каждый конкретный полиморфизм «живёт» в определенном гене, немножко изменяя свойства его продукта и, тем самым, проявление какого-то признака.
Полиморфизмы – это то, что делает нас разными. Это генетические оттенки, из-за которых один может за милую душу выпить литр молока, а другой после пары глотков будет искать туалет. Благодаря полиморфизмам у нас столько цветов глаз и волос. Из-за них у кого-то кровь сворачивается чуть быстрее среднего, а у кого-то – чуть медленнее. Удивительно, но весь этот спектр форм, цветов и особенностей задается комбинациями четырех букв-нуклеотидов, составляющих наши гены: A, G, T и C. Одну букву мы получаем от мамы, другую – от папы. Так получается наш собственный генотип: например GG, GA или TC. Результатом анализа на полиморфизмы как раз и будут пары букв. Например, в гене фактора свертывания крови V (этот ген называется F5) буквой под номером 1691 может быть G, а может – А. Отсюда три варианта генотипов: GG, GA и AA. Вариант GG — удел большинства людей, ему не свойственны какие-то особенности. Около 2-7% людей имеют генотип GА, то есть несут полиморфизм А (так называемую Лейденскую мутацию), из-за чего склонны к повышенной свертываемости крови. Людей с генотипом АА крайне мало. Грань между понятием «мутация» и «полиморфизм» тонка и неопределенна. Ученые-биологи любое отклонение от «эталона» могут называть мутацией, а врачи-практики обычно считают мутацией только то изменение, которое может приводить к болезни. Поэтому не смущайтесь, что полиморфизм в гене F5 называют Лейденской мутацией. |

Какие полиморфизмы обычно обсуждаются в контексте потери беременности?
Назовём героев этой статьи поимённо!
Не пугайтесь того, что эти названия вам ни о чем не говорят, и пока что поверьте: они и врачу вашему в большинстве случаев ничего не скажут.
- F5: 1691 G>A (Arg506Gln)NB!
- F2: 20210 G>A NB!
- F7: 10976 G>A (Arg353Gln)
- F13: G>T (Val34Leu)
- FGB: -455 G>A
- ITGA2: 807 C>T (Phe224Phe)
- ITGB3: 1565 T>C (Leu33Pro)
- SERPINE1 (PAI-1): -675 5G>4G
- MTHFR: 677 C>T (Ala222Val)
- MTHFR: 1298 A>C (Glu429Ala)
- MTR: 2756 A>G (Asp919Gly)
- MTRR: 66 A>G (lle22Met)
NB! Обратите внимание, что эти два полиморфизма могут играть важную роль в принятии решения о назначении КОК (комбинированных оральных контрацептивов).
Почему врачи назначают анализы на эти полиморфизмы?
Когда ученые узнали о существовании полиморфизмов, они задумались: а нельзя ли использовать это знание для выделения группы людей с предрасположенностью к определенным заболеваниям, и заблаговременно их предупреждать? Известно же: предупредить легче, чем лечить!
Эти времена совпали с подъемом молекулярных технологий, позволивших выполнять тесты на полиморфизмы относительно просто и недорого. Исследователи смекнули, что работы типа «Влияние полиморфизма Х на болезнь Y» генерировать легко и делать это можно практически бесконечно. Поскольку болезней и полиморфизмов много, всегда была возможность подобрать пару «полиморфизм – болезнь», позволявшую даже из безнадежных данных вытащить мало-мальски значимую связь и опубликоваться, кокетливо умолчав об изъянах дизайна исследования. Соедините немного логики и статистики – и получите скромное, но научное достижение.
Вот как рассуждали эти исследователи: уже упоминавшаяся Лейденская мутация связана с повышенной свертываемостью крови. Известно, что формирование и функционирование плаценты сильно зависит от агрегатных свойств крови, а при невынашивании беременности в плацентах нередко находят очаги тромбоза. Логично предположить, что у носительниц Лейденской мутации эти нарушения могут встречаться чаще. Осталось провести исследование и проверить эту гипотезу. Такие исследования были проведены и некоторые показали наличие связи между наличием Лейденской мутации и повышенным риском потери беременности.
Так появилась богатая (на немалую долю отечественная) «литературная база», указывающая на связь между полиморфизмами и предрасположенностью к разным болезням.
Именно на эту «базу» опирались производители реагентов при убеждении врачей в целесообразности назначения тестов на полиморфизмы. Да-да, на определенном этапе потребность в диагностикумах для анализов на полиморфизмы стала так велика, что привлекла производителей реагентов, которые создали коммерческие наборы для выполнения этих тестов. А товар требует продвижения. Как можно расширить рынок таких наборов? Внедрить тесты на полиморфизмы в клиническую практику! И эти анализы из научных лабораторий стали «заползать» в диагностические.
Когда результаты научных исследований переносятся в клиническую практику без должной оценки последствий, страдают кошельки и нервы пациентов.
Так появились лаборатории, предлагающие тесты на полиморфизмы как медицинские диагностические услуги. Так появились врачи, наученные лабораториями и производителями реагентов, что эти тесты нужно назначать в различных случаях, в том числе при невынашивании беременности. Так сформировалась целая мифология про то, какие полиморфизмы надо выявлять и как их «лечить».
Но достаточно мифов. Дальше — только факты:

1. Полиморфизмы не являются значимой причиной ранней потери беременности
Около 70% беременностей, прервавшихся в первом триместре, не могли развиваться из-за генетических аномалий ЭМБРИОНА (не матери!!!)[3]. Не путайте с генетическими полиморфизмами!
Полиморфизмы – это генетические особенности мамы, а приводящие к выкидышу нарушения структуры и количества хромосом – это грубые аномалии эмбриона. Возникновение таких эмбрионов – часть жизни, так же, как и их ранняя отбраковка.
Оставшиеся 30% ранних потерь беременности тоже не имеют отношения к полиморфизмам, а обусловлены антифосфолипидным синдромом, неправильным функционированием шейки матки, инфекциями и другими причинами, к которым генетические полиморфизмы матери не относятся.
2. Какие-то полиморфизмы есть у всех людей
В отличие от мутаций, вызывающих редкие генетические болезни, которые встречаются у одного из десятков тысяч людей, какие-то полиморфизмы есть у всех. Каждый день мимо вас проходят люди с такими же GG, GA и TC, как у вас. Возможно, у них есть дети, но может быть и нет. Есть вероятность, что они сталкивались с потерей беременности, а может быть их это несчастье обошло стороной. В любом случае: от вас они отличаются тем, что не тратили деньги на анализ полиморфизмов.
3. Полиморфизмы не определяют признак полностью (или на большую часть)
Вернемся к несчастным больным генетическими заболеваниями: их редкий генетический дефект практически на 100% определяет их беду. То, что генетики называют «факторами среды» (поведение, питание, физическая активность) вносит очень маленький вклад в их несчастье. С полиморфизмами наоборот: их вклад очень мал.
Например, вероятность развития венозного тромбоза хоть в некоторой степени и зависит от наличия, например, уже знакомой нам Лейденской мутации, но на львиную долю определяется весом, статусом курения, возрастом, наличием беременности, принимаемыми препаратами и другими факторами.
4. Полиморфизм – не болезнь
Какими бы жуткими словами не сопровождались комбинации из букв A, G, T и C в заключении генетического анализа, они НЕ говорят о том, что у женщины будет, например, «невынашивание беременности».
Пример из жизни:
Когда на бланке результата «Нарушение развития плода – незаращение нервной трубки» написано рядом с «MTRR c.66A>G G/G» любой человек поймёт такую запись как причинно-следственную связь. А это не так. Наличие полиморфизмов говорит лишь о том, что вы принадлежите к людям, у которых по данным некоторых(!) научных(!!!) исследований эти патологии возникают чаще, чем у людей без ваших полиморфизмов. И тут мы переходим к следующему факту…
5. Влияние полиморфизмов «видно» только на больших группах людей
Даже будучи специалистом, я не пойму ваш генотип по генам свертывания крови, увидев вашу коагулограмму (анализ на свертываемость крови). А всё потому, что эти различия не «видны» на индивидуальном уровне. У человека с «плохими» полиморфизмами свертывание может быть «лучше», чем у «генетически идеального». Лишь среднее значение этого показателя, измеренное в большой группе людей с «плохим» генотипом, будет отличаться от такового у группы с «хорошим».
| Немного математики: Иногда в заключении анализа рядом с жуткими «диагнозами» можно увидеть цифры. Например, «Выявленный полиморфизм в 3,5…5,5 раз увеличивает риск венозной тромбоэмболии». Эти цифры – совершенно честные[4] для Лейденской мутации. Этот полиморфизм – один из двух достойных хоть какого-то внимания полиморфизмов системы свертывания крови. Второй – так называемый «полиморфизм протромбина», c.20210G>A в гене фактора свертывания крови II (F2). Но вернемся к цифрам. Увеличение в 3,5…5,5 раз – это существенно? Конечно существенно! Если мне завтра в три с половиной раза увеличат зарплату, это будет ой как существенно… А если посмотреть не относительный, а абсолютный риск? Когда у вас есть Лейденская мутация, ваш ежегодный риск получить венозную тромбоэмболию равен 0,05…0,2%. Иными словами:
Абсолютный риск ВТЭ настолько мал, что даже увеличение в разы не делает его существенным для жизни отдельного конкретного человека. Беременность в совокупности с Лейденской мутацией повышает риск ВТЭ, но шанс на то, что тромбоза НЕ будет, всё равно не опускается ниже 95%. |
И теперь пара слов о лечении:
1. «Вылечить» полиморфизмы нельзя.
Это часть генотипа, и он останется неизменным до конца жизни. Поэтому тактика «сдать на полиморфизмы – полечить – сдать контрольный анализ» абсурдна по своей сути.
2. Ни один из полиморфизмов не является прямым поводом для назначения лечения.
Справедливости ради, стоит отметить, что при невынашивании беременности антикоагулянтная терапия может потребоваться, и она дает неплохие результаты. Но для назначения антикоагулянтов должен быть установлен диагноз «антифосфолипидный синдром» (который может сочетаться или не сочетаться с полиморфизмами в генах системы свертывания).
3. Курантил, актовегин, тромбоасс, пиявки не нужны.
Они не имеют доказанной эффективности в улучшении исходов беременности у женщин с полиморфизмами в системе свертывания.
Тестирование женщин даже с неоднократной потерей беременности на наследственные тромбофилии[5] и полиморфизмы фолатного цикла[6] не входит в рекомендации ведущих медицинских организаций, занимающихся этой проблемой. Но в большинстве отечественных «методичек» и рекомендаций по невынашиванию беременности эти исследования входят.
И чтобы не оставлять неопределенности:
Анализы на генетические полиморфизмы женщинам, столкнувшимся с потерей беременности один или несколько раз, делать не нужно
Источники:
[1]https://www.acog.org/Resources-And-Publications/Practice-Bulletins/Committee-on-Practice-Bulletins-Gynecology/Early-Pregnancy-Loss
[2] https://www.webmd.com/baby/guide/pregnancy-miscarriage#1
[3] http://emedicine.medscape.com/article/260495-overview#a11
[4] Scott M. Stevens et al. Guidance for the evaluation and treatment of hereditary and acquired thrombophilia. J Thromb Thrombolysis (2016) 41:154–164
[5] Evaluation and treatment of recurrent pregnancy loss: a committee opinion
[6] Thrombophilias and recurrent pregnancy loss: a critical appraisal of the literature
Автор: Карпачева Клавдия, молекулярный генетик
Похожее
Комментарии в Facebook
Для мутантов:) Расшифровка значения полиморфизма генов - полиморфизм гена асе - запись пользователя keisy (id1185367) в сообществе Зачатие в категории Статьи, полезная информация
Сама долго рылась - искала, чтоб по-русски было объяснено, какой ген, что означает. Вот, может, кому еще пригодится: что означают полиморфизмы генов
Генетические факторы риска привычного невынашивания беременности.
Комплекс исследования - Генетические факторы риска привычного невынашивания беременности включает в себя анализ на:
- полиморфизм G20210А гена II фактора свертываемости крови ( протромбина)
- полиморфизм G1691А гена V фактора свертываемости крови (лейденского фактора)
- полиморфизм С667Т гена метилентетрагидрофолатредуктазы ( МТНFR)
- полиморфизм 4 G/5 G гена ингибитора активатора плазминогена ( РАI-1)
- полиморфизм VaI34Leu гена фактора XIII свертываемости крови (F13)
- полиморфизм D/I гена ангиотензинпревращающего фермента ( АСЕ)
- полиморфизм А1/А2 гена CYP17 **
Привычное невынашивание беременности тесно связано с генетическими нарушениями. Риск невынашивания складывается из нескольких составляющих:
- Нарушение тромбообразования.
- Нарушение тромболизиса.
- Нарушении синтеза половых гормонов.
При повышенной склонности к тромбообразованию, нарушается система гемостаза. Генетические нарушения у женщин с наследственной тромбофилией проявляются при беременности. Предрасположенность к тромбофилии является причиной привычного невынашивания, задержки развития плода, гестозов, нарушения развития плаценты.
Генетические факторы риска.
Замедление процессов фибринолиза при мутации генов PAI -1 является причиной нарушения процесса имплантации плода. Мутация 6754/5G приводит к повышению фибрина в сосудах матки, снижению плацентарного кровообращения, что в свою очередь является причиной задержки развития плода.
Полиморфизм гена MTHFR, который является ферментом метаболизма продукта гомоцистеина. В норме это вещество не накапливается, при генетическом нарушении оно поражает сосуды и способствует образованию тромбов.
Полиморфизм гена F5 который отвечает за антикоагуляционные реакции, приводит к осложнениям беременности, невынашиванию, отставанию развития плода, поздним выкидышам, образованию тромбов в плаценте.
Мутация гена F2, который отвечает за образование протромбина, участвующего в свертывании крови, приводит к повышению его уровня в два раза. Полиморфизм этого гена является фактором всех осложнений беременности.
Ген F 13 отвечает за образование фибриназы. При мутации этого гена повышается активность фибриназы при нормальном ее количестве.
При полиморфизме гена АСЕ, отвечающего за повышение артериального давления, приводит к развитию одного из самых опасных осложнений беременности - эклампсии.
Изменения гена CYP17, отвечающего за образование стероидных гормонов, при генотипе А2/А2 и А1/А2 значительно увеличивает риск невынашивания.
Полиморфизм гена АСЕ.
Ген АСЕ участвует в превращении неактивного ангиотензина в активный. Это вещество является одним из самых активных веществ, которые повышают артериальное давление. В связи с этим могут развиваться артериальные гипертензии, эндотелиальная дисфункция, тяжелое осложнение у беременных - эклампсия.
Полиморфизм коагуляционного фактора F5 (V).
Фактор играет важную роль в регуляции свертываемости крови - образование тромбина. Мутация G1691A (мутация Лейден) приводит к гиперкоагуляции и к риску развития образования тромбов в венозных сосудах, артериальным тромбоэмболиям. Полиморфизм гена повышает риск развития коронарного стеноза, инфаркта миокарда и инсульта.
Полиморфизм коагуляционного фактора F2 (20210 G).
Коагуляционный фактор F2 (протромбин) участвует в процессах свертываемости крови (образованию кровяных сгустков). Полиморфизм 20210 G приводит к увеличению протромбина в два раза. Повышается риск возникновения тромбофилии, сердечно-сосудистым заболеваниям.
Полиморфизм гена MTHFR (С677Т).
Фактор отвечает за синтез фолиевой кислоты, а также является ферментом метаболизма гомоцистеина, который токсически действует на сосуды. Накопление гомоцистеина приводит к коронарному атеросклерозу.
Мутация гена приводит к ишемическим заболеваниям сердца, инфаркту миокарда, атеросклерозу, осложнениям беременности, дефектам развития плода.
Полиморфизм коагуляционного фактора F7 (Arg353Gln).
Фактор активирует систему свертывания крови образованию кровяного сгустка. Высокий уровень F7 повышает риск стеноза коронарных сосудов и инфаркта миокарда.
Полиморфизм тромбоцитарного рецептора фибриногена.
Фактор обеспечивает быстрое склеивание тромбоцитов и купирование поврежденного эпителия. Мутация гена приводит к повышенной агрегации тромбоцитов и образованию тромбов, что приводит к сердечно-сосудистым заболеваниям. Терапия аспирином у пациентов с мутацией данного фактора не эффективна.
Полиморфизм A фибриногена (455 G).
Фибриноген при повреждении сосудов переходит в фибрин и образует кровяные сгустки. Мутация может привести к повышенной выработке фибриногена в крови и создает высокий риск образования тромбов. Это приводит к повышенному давлению крови, инсультам и тромбоэмболическим заболеваниям. Риск инсультов при этом может увеличиться в 4 раза.
Источник тут
Е
сли забить в поисковик на этом сайте "полиморфизм", то там выскакивает много ссылок про еще разные гены, если кому-то этих не хватит))) вкратце и по-русски. нарушение в миокардеПриобретенные и наследственные факторы риска формирования тромбов
Заболевания, ассоциированные
с повышенным тромбообразованием
Антифофсфолипидный синдром
Нарушения в свертываемости крови являются одной из причин осложнений во время беременности. Имея в основе разные факторы, она сгущается и двигается по сосудам, замедляя скорость. А это в свою очередь, может вызвать образование тромбов, которые закупоривают сосуды и приводят к тяжелым последствиям.
Такие нарушения в свертываемости крови приводят к тому, что у женщины не приживается в матке плодное яйцо, происходят нарушения в плаценте, либо регулярные выкидыши на ранних сроках.
И причиной этому является антифосфолипидный синдром – это клинический иммунный симптоматический комплекс, характеризующийся тромбозами венозного и артериального характера, последствиями которого является потеря плода с неясным генезом.
Этот синдром может развиваться и имея в основе генетическую предрасположенность.
В организме человека произошло мутирование определенных генов, которые отвечают за свертывание крови. А потом эти нарушения из поколения в поколение передаются по наследству.
Чаще всего это обнаруживается при определенных ситуациях – беременность, операция, травма или прием гормонов. То есть при тех обстоятельствах, когда свертываемость крови увеличивается. Это хорошая почва на которой проявляется неблагоприятная наследственность.
Антифосфолипидный синдром при беременности провоцирует появление тромбозов в несколько раз.
Соответственно, повышается вероятность невынашивания плода. Однако это не означает, что женщинам с проблемой свертывания крови нельзя помочь. Современная медицина имеет возможность провести тщательную диагностику таких нарушений. По ее результатам можно провести отбор женщин в потенциальную группу риска. А затем приложить максимум усилий, чтобы свести на нет проявления антифосфолипидного синдрома и беременность сделать максимально безопасной, исключить возможность осложнений.
Будущей маме придется произвести корректировку образа жизни, питания и начать прием назначенных препаратов.
Прием таких лекарств необходим на протяжении всего срока беременности, особенно тем, у кого были случаи невынашивания беременности. Обязательной диагностике особенно должны подлежать женщины, если у них рождались мертвые младенцы или они вскоре умирали.
Если есть осложнения, которые связаны с антифосфолипидным синдромом, и беременность планируется в ближайшее время, в таких случаях за месяц до ее наступления необходимо начать прием противотромботических препаратов, назначенных врачом. И не прекращать лечение в течение всего периода положения беременности. Тогда шансы на благополучный исход очень велики, ведь правильное и своевременное лечение дает эффективность почти 100%!
Термин "Тромбоз" используется для обозначения появления сгустков в сосудистой системе. Чаще всего встречается тромбоз вен.
Тромбоз – серьезное заболевание, которое может привести к сердечной недостаточности (коронарный тромбоз), инсульту или гангрене, если он поразит кровеносные сосуды конечностей.
Острый тромбоз требует срочного врачебного вмешательства. Вызовите «скорую помощь» или вашего домашнего врача. Положите пациента поудобнее, обеспечьте ему тепло. Не давайте пациенту никакой еды и питья. Не разрешайте ему двигаться до тех пор, пока не придет помощь.
Никаких средств альтернативной медицины применять нельзя, пока не получено заключение врача. Длительное наблюдение за состоянием и профилактика возможного в будущем тромбоза зависит от правильного питания, физических упражнений, снижения потребления алкоголя и табака. Излишний вес также является фактором риска.
«Что такое полиморфизм генов?» – Яндекс.Кью
Можно сказать, что это будущее уже наступило, а станет общедоступным через 5-10 лет.
Наверняка вы слышали про "редактирование генома", о котором много говорят генетики последние пару лет. Речь идет о группе технологий, которые позволяют изменять ДНК у живых клеток. Пока ученые занимаются изучением возможных побочных эффектов и повышением точности таких технологий, применяя их на различных организмах, и только в редчайших случаях -- на людях.
В первую очередь, ученые надеются с помощью редактирования генома "исправлять" мутации, которые приводят к тяжелым (обычно смертельным) наследственным заболеваниям. Таким, к примеру, является мышечная дистрофия Дюшенна, которая проявляется из-за мутаций в гене DMD у каждого пятитысячного мальчика в возрасте 3-7 лет, а уже к 15 годам почти все пациенты прикованы в инвалидному креслу и вряд ли доживут до 20. Эффективного лечения у этого заболевания на данный момент нет, но 2018 году ученым удалось вылечить от такого заболевания собак: http://science.sciencemag.org/content/362/6410/86 Поэтому есть надежда, что после тщательных проверок и клинических испытаний коррекция мутаций по крайней мере в гене DMD позволит детям не болеть этим заболеванием.
Рак и многие другие заболевания тоже связаны с определенными нарушениями в нашей ДНК, поэтому развитие технологий редактирования генома позволит снизить генетические риски (например, исправлять мутации в генах BRCA1 и BRCA2, наличие которых приводит к раку молочной железы у 87% женщин). Однако рак -- это мультифакторное заболевание. Его развитие зависит не только от генов, но и вот внешних факторов, поэтому редактированием генома навсегда избавиться от рака не получится.
Однако недавно началось использование технологии, которая не исправляет, а создает специальные мутации в некоторых клетках организма. Такое направленное создание мутаций позволяет клеткам атаковать раковую опухоль в рамках иммунотерапии. За эти технологии была вручена Нобелевская премия по физиологии и медицине в 2018 году. Прочитать про иммунотерапию по-русски можно в блоге Genotek, а про редактирование генома и лечение рака -- по-английски в блоге Cancer Research UK.
Ну и уже пару десятилетий во всем мире доступна процедура ЭКО с ПГД. Это буквосочетание означает, что после оплодотворения в пробирке, сразу у нескольких эмбрионов берут буквально несколько клеток на генетический анализ. Такая процедура позволяет в ходе ЭКО отобрать только эмбрионы без мутаций, тем самым практически гарантирую родителям рождение ребенка без тяжелых заболеваний.
Но развитие таких технологий поднимает много этических вопросов. Например, для каких целей и кому нужно разрешать редактировать ДНК и исправлять мутации? Не приведет ли развитие технологий к тому, что богатые будут редактировать свою ДНК, улучшая и качество жизни и свои возможности, а у бедных не будет доступа к этой технологии, что приведет к еще большему расслоению в обществе. Можно ли разрешить парам заказывать "дизайнерских детей", то есть выбирать внешность, таланты и прочие качества? Я рекомендую прочитать доклад британского общества биоэтиков, подготовленный в 2018 году -- там поднимаются все актуальные этические вопросы применения редактирования генома и других технологий в развитии общества.
Расширенное исследование генов системы гемостаза (с описанием результатов врачом- генетиком)
Метод определения Real-time-PCR.
Исследуемый материал Цельная кровь (с ЭДТА)
Расширенное исследование генов системы гемостаза: F2, F5, MTHFR, MTR, MTRR, F13, FGB, ITGA2, ITGВ3, F7, PAI-1
Комплексное исследование генетических факторов риска развития нарушений в системе свертывания крови и фолатном цикле.
Различные изменения в генах системы гемостаза и цикла обмена фолатов предрасполагают к развитию большого числа патологических состояний: инфаркты, инсульты, тромбоэмболии, кровотечения, патология беременности и родов, осложнения послеоперационного периода и т.д.
Профиль включает в себя исследование основных полиморфизмов в генах системы гемостаза и фолатного цикла:
- F2 c.*97G>A (20210 G>A; rs1799963),
- F5 c.1601G>A (Arg534Gln; 1691 G>A; rs6025),
- MTHFR c.665C>T (Ala222Val; 677 C>T; rs1801133),
- MTHFR c.1286A>C (Glu429Ala; 1298 A>C; rs1801131),
- MTR c.2756A>G (Asp919Gly; rs1805087),
- MTRR c.66A>G (Ile22Met; rs1801394),
- F13 с.103G>T (I63Т; rs5985),
- FGB c.-467G>A (-455 G>А; rs1800790),
- ITGA2 c.759C>T (Phe253Phe, 807 C>T; rs1126643),
- ITGB3 c.176T>C (Leu59Pro; 1565 T>C; rs5918),
- F7 c.1238G>A (Arg353Gln; 10976 G>A; rs6046),
- PAI-1 (SERPINE1) –675 5G>4G (rs1799889).
Ген F2 кодирует аминокислотную последовательность белка протромбина. Полиморфизм F2 c.*97G>A приводит к повышенной экспрессии гена. Клинически неблагоприятный вариант полиморфизма (c.*97A) наследуется по аутосомно-доминантному типу. Наличие полиморфизма F2 c.*97G>A в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне курения - в 40 и более раз) увеличивает риск возникновения венозных тромбозов, в том числе тромбозов сосудов мозга и сердца, особенно в молодом возрасте. У пациентов-носителей данного полиморфизма повышен риск развития тромбоэмболий после хирургических вмешательств. Приём оральных контрацептивов у данной группы лиц также увеличивает риск тромбозов (относительный риск развития тромбофилии и венозной тромбоэмболии у гетерозиготных носительниц полиморфизма c.*97G>A возрастает в 16 раз).
Ген F5 кодирует аминокислотную последовательность белка проакцелерина - коагуляционного фактора 5. Нуклеотидная замена c.1601G>A («мутация Лейден») приводит к аминокислотной замене аргинина на глутамин в позиции 534, что придает устойчивость активной форме проакцелерина. Клинически это проявляется рецидивирующими венозными тромбозами и тромбоэмболиями. Наличие полиморфизма в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне заместительной гормонотерапии или приема оральных контрацептивов - в 30 и более раз) увеличивает риск венозных тромбозов. Риск инфаркта миокарда увеличивается в 2 и более раз, риск развития патологии беременности (прерывание беременности, преэклампсия, хроническая плацентарная недостаточность и синдром задержки роста плода) увеличивается в 3 и более раз.
Также, пациенты, являющиеся одновременно носителями полиморфизма c.*97G>A гена протромбина и «мутации Лейден», еще в большей степени подвержены риску развития тромбозов и тромбоэмболий.
Ген MTHFR кодирует аминокислотную последовательность фермента метилентетрагидрофолатредуктазы, играющего ключевую роль в метаболизме фолиевой кислоты. Полиморфизм c.665C>T гена MTHFR связан с заменой нуклеотида цитозина (С) на тимин (Т), что приводит к аминокислотной замене аланина на валин в позиции 222. Вариант c.665Т связан с четырьмя группами мультифакториальных заболеваний: сердечно-сосудистыми, дефектами развития плода, колоректальной аденомой и раком молочной железы и яичников. У женщин с генотипом c.665Т/Т дефицит фолиевой кислоты во время беременности может приводить к порокам развития плода, в том числе незаращению нервной трубки. Неблагоприятное воздействие варианта c.665Т- зависит от внешних факторов: низкого содержания в пище фолатов, курения, приема алкоголя. Сочетание генотипа c.665Т/Т и папилломавирусной инфекции увеличивает риск цервикальной дисплазии. Назначение препаратов фолиевой кислоты может значительно снизить негативное влияние данного варианта полиморфизма.
Полиморфизм MTHFR c.1286A>C связан с точечной заменой нуклеотида аденина (А) на цитозин (С), что приводит к замене аминокислотного остатка глутаминовой кислоты на аланин в позиции 429, относящейся к регулирующей области молекулы фермента. При наличии данного полиморфизма отмечается снижение активности фермента MTHFR. Это снижение обычно не сопровождается изменением уровня гомоцистеина в плазме крови у носителей дикого варианта полиморфизма c.665C>T, однако сочетание аллельного варианта* c.1286C с аллелем c.665T приводит к снижению уровня фолиевой кислоты и соответствует по своему эффекту гомозиготному состоянию MTHFR c.665Т/T. При этом риск развития дефектов нервной трубки повышается в 2 раза. Жизнеспособность плодов, имеющих одновременно оба полиморфных варианта, также снижена.
Ген MTR кодирует аминокислотную последовательность фермента метионин синтазы. Полиморфизм c.2756A>G связан с аминокислотной заменой (аспарагиновой кислоты на глицин) в молекуле фермента. В результате этой замены функциональная активность фермента изменяется, что приводит к повышению риска формирования пороков развития у плода. Влияние полиморфизма усугубляется повышенным уровнем гомоцистеина.
Ген MTRR кодирует аминокислотную последовательность фермента редуктазы метионинсинтазы. Полиморфизм c.66A>G связан с аминокислотной заменой в молекуле фермента. В результате этой замены функциональная активность фермента снижается, что приводит к повышению риска развития дефектов нервной трубки у плода. Влияние полиморфизма усугубляется дефицитом витамина В12. При сочетании полиморфизма c.66A>G гена MTRR с полиморфизмом c.665C>T в гене MTHFR риск spina bifida увеличивается. Полиморфизм c.66A>G гена MTRR усиливает гипергомоцистеинемию, вызываемую полиморфизмом c.665C>T в гене MTHFR.
Ген фибриназы (F13) кодирует синтез трансглютаминазы, участвующей в стабилизации фибринового сгустка и в формировании соединительной ткани. Аллельные варианты с.103G/Т и с.103Т/Т приводят к снижению уровня трансглютаминазы с образованием сетчатой структуры фибрина с более тонкими волокнами, меньшими порами, и изменением характеристик проникновения, которое в сочетании с другими факторами риска ассоциируется с возможным риском внутричерепных кровоизлияний и кровотечений из внутренних органов, а также привычным невынашиванием беременности. При этом аллельный вариант с.103Т может выступать в роли протективного фактора в отношении инфаркта миокарда и венозных тромбозов.
Ген FGB кодирует β-цепь фибриногена, являющегося предшественником фибрина. Аллельный вариант c.-467А обусловливает усиленную транскрипцию гена и может приводить к увеличению уровня фибриногена в крови и повышению вероятности образования тромбов при наличии дополнительных факторов риска. Гетерозиготный вариант c.-467G/А связывают с повышенным риском ишемического инсульта и лакунарными инфарктами церебральных сосудов. Гомозиготный вариант c.-467A/А связывают с повышенным риском инфаркта миокарда.
Ген гликопротеина Gp1a (ITGA2) кодирует синтез альфа-2-субъединицы интегринов – специализированных рецепторов тромбоцитов. Аллельный вариант c.759Т вызывает изменение первичной структуры субъединицы и свойств рецепторов. При гетерозиготном (c.759C/T) варианте отмечается увеличение скорости адгезии тромбоцитов к коллагену I типа, что может приводить к повышенному риску тромбофилии, инфаркта миокарда и других сердечно-сосудистых заболеваний. Аллельный вариант c.759Т связывают со случаями резистентности к аспирину. Помимо этого, при гомозиготном (c.759Т/T) варианте значительно увеличивается количество рецепторов на поверхности тромбоцитов. В совокупности, при гомозиготном варианте данного полиморфизма значительно повышен риск тромбофилии, инфаркта миокарда и развития других острых эпизодов тромбообразования в возрасте до 50 лет, даже по сравнению с гетерозиготным вариантом.
Ген гликопротеина Gp3a (ITGB3) кодирует синтез бета-3 цепи интегринового комплекса GP2b\3a, участвующего в разнообразных межклеточных взаимодействиях (адгезии и сигнализации).
Аллельный вариант c.176С (гетерозигота c.176T/C) обусловливает повышенную адгезию тромбоцитов и может приводить к увеличению риска развития острого коронарного синдрома, а также связан с синдромом привычного невынашивания беременности. Гомозиготный вариант c.176С/C обусловливает повышенную адгезию тромбоцитов и может приводить к значительному увеличению риска развития острого коронарного синдрома в возрасте до 50 лет. У лиц с полиморфными аллельными вариантами часто отмечается пониженная эффективность аспирина.
Аллельный вариант c.1238A (гетерозигота c.1238G/A и гомозигота c.1238А/A) гена F7 приводит к понижению экспрессии гена и снижению уровня фактора 7 в крови, рассматривается как протективный маркёр в отношении развития тромбозов и инфаркта миокарда.
Ген ингибитора активатора плазминогена (PAI-1) кодирует белок-антагонист тканевого и урокиназного активатора плазминогена. Преобладающим в популяции вариантом исследуемого полиморфизма является гетерозиготный вариант -675 5G/4G. В связи с этим данный полиморфизм самостоятельного диагностического значения не имеет, эффект возможно оценить в сочетании с другими факторами предрасполагающими к развитию патологии (например в сочетании с FGB c.-467A). Аллельный вариант -675 4G сопровождается большей активностью гена, чем -675 5G, что обусловливает более высокую концентрацию PAI-1 и уменьшение активности противосвёртывающей системы. Гомозигота -675 4G/4G ассоциирована с повышением риска тромбообразования, преэклампсии, нарушением функции плаценты и самопроизвольного прерывания беременности.
*Примечание: иногда в научной литературе при описании однонуклеотидных замен, характерных для генных полиморфизмов, встречается термин "мутантный аллель". Это терминологическая неточность, так как в классической генетике термин "мутантный аллель" традиционно рассматривается как синоним термина "мутация". При мутациях, как известно, изменение структуры гена приводит к образованию (экспрессии) нефункциональных белков и к неизбежному развитию наследственного заболевания. При полиморфизмах изменение в структуре гена приводит лишь к появлению белков с немного изменёнными физико-химическими свойствами. Такие изменения, как известно, проявляют себя при воздействии на организм различных факторов внешней среды или при изменении функционального состояния организма человека. И только в таких ситуациях функционирование белков со структурными особенностями может, либо способствовать ускорению развития заболевания, либо, напротив, тормозить формирование патологических процессов. Поэтому, на наш взгляд, для разграничения изменений в генах столь очень похожих структурно, но приводящих к несоизмеримо разным последствиям для организма, корректнее в отношении генных полиморфизмов применять понятие "аллельный вариант гена", а не "мутантный аллель".
За генный полиморфизм приходится платить
Статья на конкурс «био/мол/текст»: В меняющихся условиях окружающей среды генетическое разнообразие в популяции дает отдельным особям преимущество. Оно не только позволяет виду выживать и существовать в геологических масштабах времени, но и является основой генетической изменчивости и непрерывного процесса видообразования. Обычно белки, кодируемые разными аллелями одного гена, обладают одинаковыми функциональными свойствами, и само по себе наличие генных полиморфизмов не оказывает никакого влияния на жизнедеятельность организма, являясь выражением его биологической индивидуальности. Но эти генетические различия вносят важный вклад в индивидуальные особенности развития защитных реакций и предрасположенность к целому ряду заболеваний, среди которых — невынашивание беременности.
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2013 в номинации «Своя работа».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
Генетическое разнообразие — добро или зло?
Бурное развитие геномики, происходящее в последнее время [1], предоставляет новые мощные инструменты описания и анализа генетического разнообразия. Как правило, гены представлены в популяции двумя или более вариантами — аллелями, — отличающимися между собой либо одиночными заменами нуклеотидов (SNP — single nucleotide polymorphism), то есть реальными изменениями «генетического кода», либо числом повторяющихся фрагментов ДНК. Именно SNP особенно важны для молекулярной диагностики болезней.
Что нового в понимании генетической вариабельности человека и в приложении к персонифицированной медицине дают персональные геномы? Во-первых, открываются новые генетические варианты — в каждом человеческом геноме обнаруживается порядка 3 млн. SNP на 3 млрд. нуклеотидов (это примерный размер генома целиком), что дает уровень различий один нуклеотид на 1000 п.н. Геномы двух разных людей пересекаются примерно по половине SNP. Иногда замена всего одного нуклеотида может стать причиной заболевания, но чаще всего сочетание нескольких генетических полиморфизмов может либо предрасполагать, либо препятствовать проявлению различных заболеваний, и тогда из называют мультифакториальными (МФЗ).
К мультифакториальным заболеваниям относится подавляющее большинство хронических болезней человека, включая сердечнососудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Полиморфизмы «генов предрасположенности» в сочетании с неблагоприятными внешними факторами (нерациональное питание, вредные привычки, загрязнения окружающей среды, инфекции) повышают риск развития заболевания.
В ходе эволюции SNP появляются в результате мутации единого аллеля-предшественника. Такое огромное количество изменений в жизненно-важной генетической программе возникает потому, что в ДНК любой клетки человека ежедневно происходят тысячи случайных изменений (например, депуринизация — отрыв основания аденина или гуанина от нуклеотида), а в процессе репликации самым частым классом ошибок являются замены одиночных пар нуклеотидов. Замена нуклеотида в последовательности ДНК может произойти в регулирующей области гена и значительно изменить уровень его экспрессии (количество транскрибируемой РНК), либо в кодирующей области, отвечающей непосредственно за «считываемый» с гена белок-продукт. А может быть и такой случай, что ДНК мутирует, а продукт гена остается неизменным — в этом случае говорят о синонимичной замене.
Скорость мутации нуклеотидной пары равна примерно 10−8 на поколение. Зная число пар нуклеотидов в геноме (3 млрд) можно вычислить, что каждая гамета содержит примерно 30 однонуклеотидных замен, т.е. каждый ребенок в мире рождается с примерно 60 новыми SNP! Изменение одной «буквы» в гене может существенно повлиять на работу белка-продукта за счет изменения пространственной организации белковых доменов (рис. 1), либо за счет нарушения посттранскрипционных изменений РНК (сплайсинга и редактирования) или посттрансляционных модификаций белка (процессинга и присоединения различных химических групп к аминокислотным остаткам). А может и не повлиять. Такие несинонимичные замены (nSNVs — nonsynonymous single nucleotide variants) с точки зрения эволюции считаются нейтральными (ни вредными, ни полезными). Большая часть SNP, конечно же, относится именно к этому классу, иначе рождаемые дети просто-напросто не выживали бы.
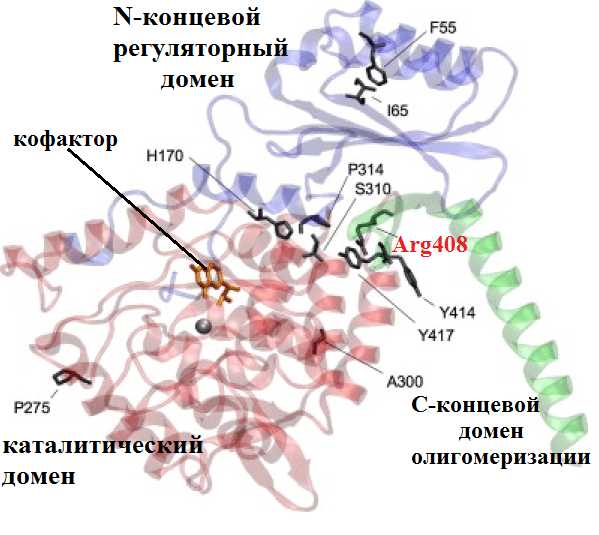
Рисунок 1. Вредные несинонимичные замены. Пример такой замены — полиморфизм PAH R408W, причина довольно известного заболевания фенилкетонурии (ФКУ). На этикетках газированных напитков и жевательных резинок можно встретить надпись «не рекомендовано больным фенилкетонурией». А все потому, что эти продукты содержит подсластитель аспартам, который состоит из двух аминокислот — аспарагиновой кислоты и фенилаланина. Организм здорового человека последнюю перерабатывает, однако у больных ФКУ — редким врожденным заболеванием (оно диагностируется у одного из 10 000 новорожденных) — фенилаланин и продукты его распада накапливаются в организме и могут привести к поражению мозга. Больных детей до совершеннолетия нужно кормить пищей, не содержащей фенилаланина. На молекулярном уровне это объясняется тем, что в гене фенилаланин-4-гидроксилазы в 12-м экзоне происходит замена цитозина на тимин. Вследствие этого возникает замена аргинина на триптофан в 408 положении аминокислотной последовательности печеночного фермента, который в норме катализирует превращение фенилаланина в тирозин. В результате замены нарушается сборка активного белка («портится» взаимодействие между двумя субъединицами тетрамера), и фермент не может больше выполнять свои функции. На рисунке изображена доменная структура олигомера гидроксилазы и обнаруженные SNP, нарушающие фолдинг белка [1].
Один ген может иметь много полиморфных вариантов. Большинство замен в них является безвредными (нейтральными). Наиболее часто встречаемый аллель называют нормальным, а редкие варианты — мутантными. Если при конкретном заболевании наблюдается более высокая частота определенного аллельного варианта (неважно — мутантного или нормального), то такой SNP называют ассоциированным с заболеванием, а полиморфный ген — геном-кандидатом предрасположенности к развитию МФЗ. Развитие МФЗ может быть запущено либо одной причиной, либо комбинацией нескольких, которые могут быть чисто генетическими (например, один или несколько аллельных вариантов комплекса генов), либо чисто средовыми (например, химические аллергены, действию которых подвергается работник лакокрасочного завода или парикмахер), либо поведенческими (например, пристрастие к определенной пище), либо социальными или психологическими. При этом индивидуальный вклад каждой причины в проявление болезни может быть незначительным, и только их сумма ведет к развитию заболевания.
Пример МФЗ, к развитию которого приводит только наличие определенных комбинаций аллельных вариантов в генах предрасположенности — невынашивание беременности (НБ) (рис. 2). Сочетание аллельных вариантов с действием факторов среды пока мало изучено, однако на данный момент в мире исследованы SNP более 90 генов, относящихся к генной сети патологии беременности. Большая часть работ посвящена исследованию генов системы детоксикации, фолатного обмена, факторов свертывания крови, HLA-системы, факторов роста, генов антиоксидантной защиты и генов, вовлеченных в воспалительные процессы.
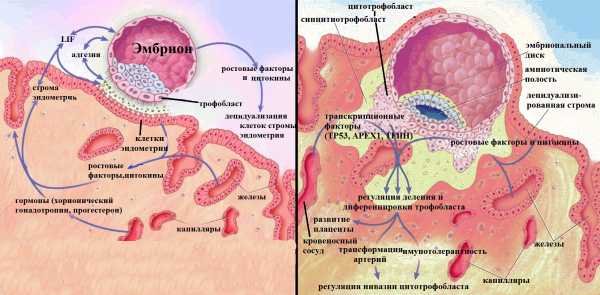
Рисунок 2. Имплантация эмбриона и причины невынашивания. Успех беременности зависит от полноценной имплантации эмбриона в организм матери. Успешная имплантация — результат сложных взаимодействий между гормонально подготовленным эндометрием матки и зрелой бластоцистой. Имплантация бластоцисты в эндометрий включает два этапа: 1) адгезия двух клеточных систем — эндометрия и трофобласта — и 2) децидуализация стромы эндометрия. Затем начинается фаза внедрения трофобласта в стенку эндометрия и активная дифференцировка трофобласта. Активно делящиеся клетки цитотрофобласта, сливаясь, образуют путем эндоредупликации синцитиотрофобласт, напрямую контактирующий с материнской кровеносной системой. В этом процессе активно задействованы все молекулы-участники регуляции и контроля клеточного цикла. Цитотрофобласт внедряется в децидуа и изменяет кровеносные сосуды матери для обеспечения тока крови к плоду: образуются хорион и плацента. Если при формировании плаценты инвазия трофобласта будет недостаточной, то произойдет выкидыш или задержка развития плода [7].
На базе кафедры генетики и НИИ Биологии ЮФУ мы изучали ассоциацию SNP генов, отвечающих за контроль повреждений ДНК, c невынашиванием беременности [5]. Эти гены называются DDR — DNA damage response, — куда относятся гены системы репарации и контроля клеточного цикла, о которых более подробно будет сказано позже.
Большие перемены — результат накопления небольших изменений
Почему именно эти гены были выбраны для исследования? Все объясняется тем, что в начале развития эмбрион должен внедриться в стенку матки. Это ключевой момент для успешного протекания беременности. При этом клетки трофобласта и стромы эндометрия делятся не традиционным образом, а путем эндоредупликации (удвоения хромосом без деления), невозможной без участия белков, контролирующих клеточный цикл и репарацию (рис. 3).
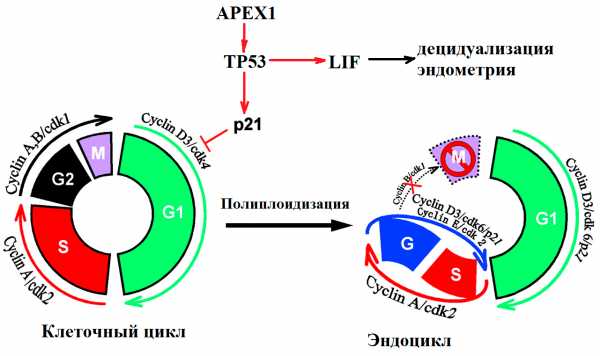
Рисунок 3. Участие генов системы репарации и контроля клеточного цикла в подготовке к имплантации эмбриона. Процесс образования специализированных клеток децидуа — ключевой для развития беременности. В начале децидуализации стромальные клетки, непосредственно окружающие имплантируемую бластоцисту, активно делятся. Клеточный цикл осуществляется за счет работы циклин-зависимых киназ. Во время деления клеток эндометрия белок p21 подавляет работу комплекса циклина D и киназы cdk4, что приводит к образованию эндоцикла (удвоению хромосом без деления). В результате эндоредупликации получаются гигантские полиплоидные клетки децидуа (до 64n). На рисунке показана роль регуляторов клеточного цикла в «обычном» митозе и в эндоцикле клеток стромы эндометрия и трофобласта [8–10].
Несмотря на то, что под воздействием неблагоприятных факторов внешней среды, а также во время процесса эндоредупликации происходит огромное количество ошибок, за год в каждой клетке накапливается очень небольшое число стабильных изменений нуклеотидной последовательности. Среди множества случайных замен оснований в ДНК лишь одна на тысячу приводит к возникновению мутации. Все остальные повреждения очень эффективно ликвидируются в процессе репарации ДНК (рис. 4).
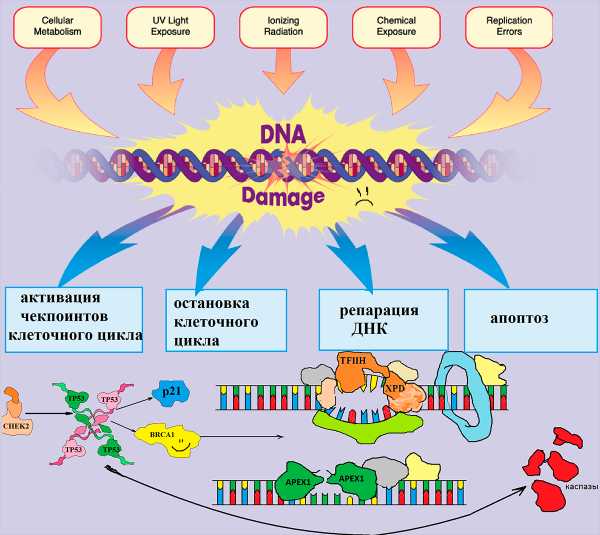
Рисунок 4. Механизм работы системы контроля повреждений ДНК. В раннем эмбриональном периоде происходит активное деление как клеток плода, так и материнских клеток. Этот процесс осуществляется ансамблем слаженно действующей системы репарации. Излучения (радиация, УФ), побочные продукты метаболизма, которые образуются внутри клеток, токсины, попадающие в организм с воздухом, пищей или питьем, а также ошибки репликации вызывают изменения в нуклеотидной последовательности ДНК, которые в норме узнаются белками-участниками контроля клеточного цикла.
Среди последних — белковые продукты генов CHEK2 и TP53, которые останавливают деление клеток и являются объектом нашего исследования. После того, как они срабатывают, ошибка исправляется белками репарации, в числе которых — белковые продукты генов APEX1 и XPD, также ставшие объектами нашей работы. Если же ошибку исправить не удастся, то включается механизм «самоубийства» клетки (апоптоза). Всё это вместе составляет сложную сеть взаимодействующих сигнальных каскадов в клетке, изменение работы одного из которых может привести к нарушению работы и гибели клетки.
Показано, что при различных осложнениях беременности наблюдается повышенный уровень апоптоза в трофобласте [4]. В результате происходит недостаточная подготовка стенок матки к имплантации эмбриона и изменения в маточно-плацентарных артериях, что приводит к снижению кровотока в них. Следствием этого является гипоксия, запускающая механизм окислительного стресса. Гипоксия усиливает апоптоз в трофобласте путем повышения экспрессии ТР53.
Именно поэтому для своих исследований мы выбрали гены системы репарации и контроля клеточного цикла. Исследование генов, контролирующих процесс репарации, — одна из важных задач в раскрытии патогенетических звеньев имплантации эмбриона и децидуализации эндометрия. Знание их роли в патогенезе невынашивания беременности позволяет, с одной стороны, прогнозировать риск развития патологии или тяжесть ее течения, с другой — индивидуально подобрать специфическую терапию для конкретного пациента.
История одного исследования
Участницы нашего исследования были разделены на две группы: женщины с нормально протекавшей беременностью, решившие прервать данную беременность на сроке 6–12 недель, и женщины с НБ. Из тканей матери (периферическая кровь и эндометрий) и плода (хорион, эмбрион) выделяли ДНК и изучали геном испытуемых на наличие аллельных вариантов Asp148Glu, Lys751Gln, 1100delC и Pro72Arg генов APEX1, XPD, CHEK2 и TP53, соответственно. Всё это — гены системы контроля клеточного цикла и репарации ДНК.
Анализ экспрессии этих генов позволил выявить возможные причины и механизмы нарушения процессов эмбриогенеза, приводящих к развитию патологии беременности. В ходе исследования мы обнаружили, что при одновременном наличии полиморфных аллелей изучаемых генов (как в генотипе плода, так и в генотипе матери) достоверно повышается вероятность развития патологии беременности. Большая часть полученных нами отличий в частоте полиморфных аллелей между исследуемыми группами выявлена именно при анализе фетальной ткани (хорион), что говорит о значительном вкладе отцовского генотипа в нормальное развитие беременности.
Это объясняется тем, что процесс имплантации эмбриона контролируется сложной молекулярной машиной, руководимой белками-участниками систем репарации и контроля клеточного цикла (рис. 5). Во время активного деления клеток неизбежно возникновение ошибок. Эти ошибки контролируются в специальных «сверочных точках» (чекпоинтах) клеточного цикла. Белки-сенсоры обнаруживают повреждения ДНК и активируют белки-датчики, которые, в свою очередь, активируют белки-эффекторы. Одним из таких белков является CHEK2 (чекпоинт-киназа2). Киназа активирует опухолевый супрессор TP53, известный также как «страж генома». Этот белок подавляет развитие многих типов новообразований, стимулирует остановку клеточного цикла и репарацию или апоптоз (в зависимости от физиологических условий и типа клеток). Действуя в качестве транскрипционного фактора, TP53 связывается со специфическими последовательностями ДНК и активирует большое число генов-мишеней. Известно, например, что TP53 избирательно индуцирует экспрессию гена хорионического гонадотропина человека (ХГЧ). ХГЧ — один из самых ранних формируемых эмбрионом сигналов, который обеспечивает поддержание продукции прогестерона и иммунную толерантность материнских тканей к ткани плода (являющейся наполовину чужеродной для организма матери из-за наличия отцовского генома).
ТР53 вызывает остановку клеточного цикла, активирует репарацию и транскрипцию необходимых для этого процесса генов (XPD, APEX1). APEX1 — эндонуклеаза, разрезающая ДНК в депуринизованных участках, способствуя процессам репарации. Кроме того, она может выступать в качестве транскрипционного фактора, активирующего ТР53 в ответ на окислительный стресс. Ген XPD кодирует белок, носящий название «хеликазная XPD субъединица основного комплекса транскрипционного фактора TFIIH». (Хеликаза — это белок, «расплетающий» двойную спираль во время репликации.) В случае успешной репарации ход клеточного цикла возобновляется, в противном же случае ТР53 запускает механизм апоптоза.
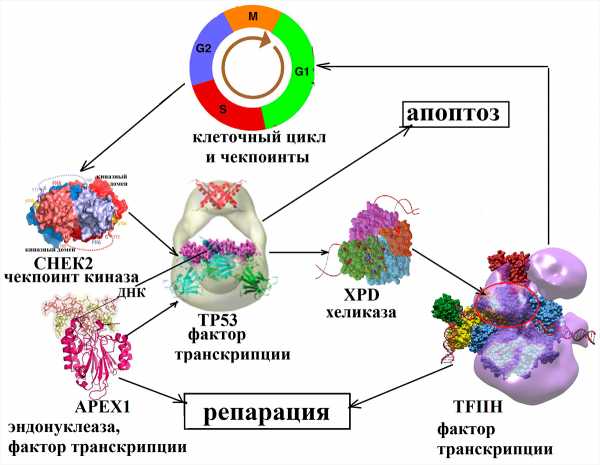
Рисунок 5. Взаимодействие белков репарации и контроля клеточного цикла. В ответ на повреждение ДНК чекпоинт-киназа активирует опухолевый супрессор, который вызывает остановку клеточного цикла и активирует хеликазу и эндонуклеазу, осуществляющие репарацию.
В результате SNP гена в молекуле его белкового продукта одна аминокислота может быть заменена на другую, что приводит к изменению конформации, размера, заряда и других свойств белка. Часто эти изменения приводят к изменению активности (как в случае с TP53, рис. 6) или потере функции (как в случае с CHEK2, рис. 7).
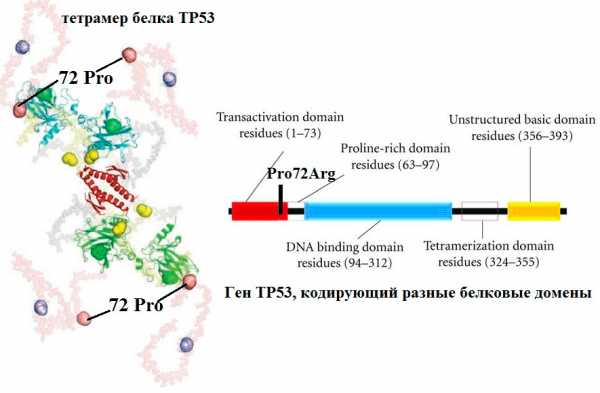
Рисунок 6. Повышение активности белка опухолевого супрессора TP53 в результате SNP кодирующего его гена. Полиморфизм Pro72Arg белка TP53 расположен в участке, богатом пролином и отвечающем за взаимодействие ингибитором TP53. Ингибитор связывается с Pro-формой гораздо сильнее, чем с Arg-формой, поэтому TP53/Arg72 эффективнее активирует апоптоз, чем исходная форма.
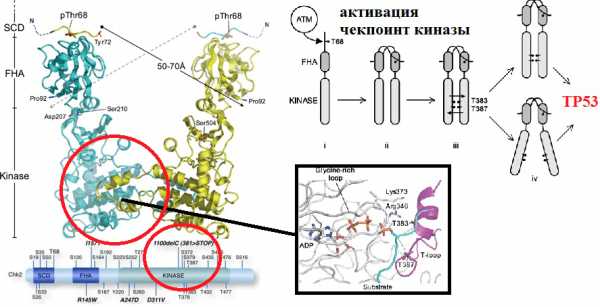
Рисунок 7. Потеря белком киназной функции в результате SNP в кодирующем его гене. Полиморфизм 1100delC гена CHEK2 приводит к экспрессии укороченного киназного домена. Это приводит к тому, что активация белка в ответ на повреждения ДНК становится невозможной (для нее необходимы 387 и 383 остатки треонина, а длина мутантного белка лишь 381 остаток из-за делеции).
При случайном одновременном наличии нескольких аллельных вариантов, продукты генов которых задействованы в выполнении схожих функций, может произойти нарушение работы всего метаболического пути и клетки в целом. Так, полиморфизм Lys751Gln гена XPD нарушает связывание с транскрипционным фактором (ТФ) IIH, действующим на ядерные рецепторы и обеспечивающим контроль активации генов гормонального ответа, необходимый для нормального развития плаценты. Вследствие этого в сыворотке крови матери повышается уровень ХГЧ (хорионического гонадотропина человека) и нарушается функция плаценты. SNP попадает в домен, богатый пролином, необходимый белку для полной индукции апоптоза. В случае неэффективной имплантации эмбриона и возникновения гипоксии будут нарушены механизмы апоптоза. Кроме того, ТР53 не сможет эффективно активировать необходимый для имплантации регулятор LIF и белок р21, запускающий процесс эндоредупликации в эндометрии, а также ХГЧ, участвующий в плацентации посредством стимуляции роста сосудов.
Полиморфные варианты генов системы репарации и контроля клеточного цикла производят менее активные белки репарации и специфические протеинкиназы, что вносит немалый вклад в развитие патологии беременности вследствие несрабатывания механизма корректировки повреждений ДНК, появления геномной нестабильности и запуска апоптоза в материнских и фетальных клетках (клетках плода), что подтвердилось в ходе нашего исследования.
По ту сторону добра и зла
Особенности спектров генетических полиморфизмов в зависимости от географических условий, диеты и этнической принадлежности говорят о невидимой руке естественного отбора. Имеются исследования, в которых было установлено, что частота аллеля Pro гена ТР53 тесно связана с географической широтой и температурой: она гораздо выше в экваториальных популяциях (рис. 8) [6].
Рисунок 8. Предполагаемый механизм отбора полиморфизма гена TP53. Под влиянием более низких температур в популяции отбирается TP53 Arg72 аллель, так как он дает более активную форму TP53, повышающего активность белков SCO2 (участвует в синтезе АТФ) и TIGAR (участвует в регуляции гликолиза и защите от окислительного стресса). TP53 Arg72 аллель участвует в регуляции метаболизма и имплантации эмбриона и повышении уровня приспособленности.
Эти факты особенно интересны в свете нейтральной теории молекулярной эволюции (НТМЭ), которая объясняет происхождение и распределение nSNV, ассоциированных с болезнями. Авторы одного из недавних исследований предполагают, что большинство аллельных вариантов, ассоциированных с заболеваниями, в современных человеческих популяциях изначально были нейтральными, а некоторые из них могли иметь адаптивное значение (например, замена глутамина на валин в одной из цепей гемоглобина — причина серповидно-клеточной анемии). В рамках этой теории nSNV, ассоциированные с МФЗ, имеют равную скорость эволюции с нейтральными nSNV. Это объясняется тем, что на них, в отличие от nSNV, приводящих к развитию моногенных болезней (например, фенилкетонурии), не действует очищающий отбор, элиминирующий заболевших индивидов [3]. Этот подход хорошо объясняет то, что исследуемый нами полиморфизм Asp148Glu гена АРЕХ1 является исключительно нейтральным для функционирования белка — он не теряет своей эндонуклеазной активности [11], — однако нами выявлена достоверная связь между наличием этого SNP в генотипе матери или плода и невынашиванием беременности.
Таким образом, НТМЭ обеспечивает клиническую генетику информативной базой для объективной оценки адаптивных событий человеческой истории как одной из основных причин заболеваний человека.
Заключение
Индивидуальный вклад каждой причины в проявление болезни может быть незначительным, и только их суммы может оказаться достаточно для развития заболевания. Именно поэтому очень сложно определить, какой именно фактор послужил сигналом для запуска патогенеза. С точки зрения индивидуального прогноза здоровья и оценки риска МФЗ каждый персональный геном дает исчерпывающую информацию о носительстве аллелей, связанных с клиническими фенотипами. Ярким примером МФЗ является невынашивание беременности (НБ). Частота невынашивания в первые три месяца беременности может достигать 80%, и, по меньшей мере, половина этих случаев происходит по непонятным причинам.
Интеграция геномики и феномики в рамках системной биологии, появившиеся в последнее время новые мощные инструменты описания и анализа генетического разнообразия — секвенирование индивидуальных геномов [12] и полногеномный анализ SNP на биочипах, проекты HapMap и «1000 геномов» [1] — дают надежду на быстрый прогресс в каталогизации генетического разнообразия, связанного с риском развития распространенных болезней. Эта информация повысит эффективность лекарственной терапии и перебросит мостик от фундаментальных научных исследований к доказательным рекомендациям в персонифицированной медицине.
Молекулярно-генетическая диагностика позволит врачу заглянуть в индивидуальную программу жизни человека, увидеть особенности его организма, предрасположенность к одним заболеваниям и устойчивость к другим. Таким образом, диагностика заболеваний на досимптомном этапе развития [13] позволит своевременно провести адекватную профилактику заболевания (например, невынашивания беременности или сахарного диабета) и назначить индивидуальную, подходящую именно для этого пациента схему лечения.
Существование генных полиморфизмов является результатом действия факторов микроэволюции и вносит вклад в генетическое разнообразие популяции, тем самым обеспечивая их удивительной способностью изменяться в соответствии с бесконечной изменчивостью окружающего мира. Исследование и выявление генных полиморфизмов, вносящих вклад в развитие того или иного заболевания, имеет прямую прогностическую ценность. Именно такой подход к терапии позволит свести к минимуму неблагоприятные эффекты лекарственных препаратов, сохранить пациенту здоровье и даже жизнь.
- Перевалило за тысячу: третья фаза геномики человека;
- Søren W. Gersting, Kristina F. Kemter, Michael Staudigl, Dunja D. Messing, Marta K. Danecka, et. al.. (2008). Loss of Function in Phenylketonuria Is Caused by Impaired Molecular Motions and Conformational Instability. The American Journal of Human Genetics. 83, 5-17;
- J. T. Dudley, Y. Kim, L. Liu, G. J. Markov, K. Gerold, et. al.. (2012). Human genomic disease variants: A neutral evolutionary explanation. Genome Research. 22, 1383-1394;
- Щербаков В.И. (2011). Апоптоз в трофобласте и его роль при патологии беременности. «Успехи современной биологии». 2, 145–158;
- Куцын К.А., Коваленко К.А., Машкина Е.В., Шкурат Т.П. (2013). Молекулярно-генетический анализ полиморфизмов генов системы репарации и контроля клеточного цикла у женщин с невынашиванием беременности. «Современные проблемы науки и образования». 1;
- Hong Shi, Si-jie Tan, Hua Zhong, Wenwei Hu, Arnold Levine, et. al.. (2009). Winter Temperature and UV Are Tightly Linked to Genetic Changes in the p53 Tumor Suppressor Pathway in Eastern Asia. The American Journal of Human Genetics. 84, 534-541;
- Errol R. Norwitz, Danny J. Schust, Susan J. Fisher. (2001). Implantation and the Survival of Early Pregnancy. N Engl J Med. 345, 1400-1408;
- S. K. Dey, H. Lim, Sanjoy K. Das, Jeff Reese, B. C. Paria, et. al.. (2004). Molecular Cues to Implantation. Endocrine Reviews. 25, 341-373;
- Gianluca Tell, Franco Quadrifoglio, Claudio Tiribelli, Mark R. Kelley. (2009). The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxidants & Redox Signaling. 11, 601-619;
- W. Hu, T. Zheng, J. Wang. (2011). Regulation of Fertility by the p53 Family Members. Genes & Cancer. 2, 420-430;
- David M. Wilson, Daemyung Kim, Brian R. Berquist, Alice J. Sigurdson. (2011). Variation in base excision repair capacity. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 711, 100-112;
Скверный анекдот: негр, китаец и Крейг Вентер...;- Упреки в нарциссомике.
Клиническое значение выявления полиморфизмов в геноме человека
Система свертываемости крови
Полиморфизмы генов системы свертываемости крови
Мутация Лейден 1691 G>A коагуляционного фактора V (F5)
Физиология и генетика. Коагуляционный фактор V или фактор V свертывания крови является белковым кофактором при образовании тромбина из протромбина. Полиморфизм G1691A Leiden (аминокислотная замена Arg (R) -> Gln (Q) в позиции 506, известная также как «мутация Лейден» или «Ляйден») является показателем риска развития венозных тромбозов. Это точечная (однонуклеотидная) мутация гена, кодирующего фактор V свертывания крови, придает устойчивость активной форме фактора V к расщепляющему действию специализированного регулирующего фермента, С-белка, что приводит к гиперкоагуляции. Соответственно, риск образования тромбов повышается.Распространенность мутации в популяциях европейского типа составляет 2-6%.
Показания к анализу. Венозный тромбоз, развитие тромбоэмболических заболеваний в молодом возрасте; рецидивирующий характер тромбоэмболизмов; сердечно-сосудистые заболевания в семейном анамнезе, заместительная гормонотерапия, прием гормональных контрацептивов, невынашивание беременности, фетоплацентарная недостаточность, внутриутробная гибель плода, токсикоз, задержка развития плода, отслойка плаценты, пациентам, готовящимся к большим полостным операциям (миома матки, кесарево сечение, кисты яичников и пр.).
Клинические данные. Наличие мутации Лейден повышает вероятность развития целого ряда осложнений беременности: выкидыш на ранних сроках (риск повышается в 3 раза), отставания развития плода, позднего токсикоза (гестоза), фетоплацентарной недостаточности. Повышенная склонность к тромбообразованию может приводить к артериальным тромбоэмболиям, инфаркту миокарда и инсульту. Наличие мутации Лейден повышает риск первичных и рецидивирующих венозных тромбозов, по крайней мере, в 3-6 раз. Приводимые ниже примеры иллюстрируют связь мутации с различными видами тромбозов и другими кардиоваскулярными заболеваниями.
В течение 8 лет в нескольких центрах проводилось исследование более 300 пациентов с венозной тромбоэмболией (ВТЭ), в ходе которого был установлен повышенный 3,7-кратный риск ВТЭ при наличии мутации Лейден [11929758]. В другой работе пациенты с венозной тромбоэмболией исследовались в течение 68 месяцев. За это время 14% пациентов перенесли рецидив ВТЭ. Мутация Лейден фактора V приводит к четырехкратному увеличению риска повторного ВТЭ. Для пациентов с ВТЭ, имеющим мутацию Лейден, рекомендована более длительная антикоагуляционная терапия по сравнению с пациентами с нормальным фактором V ([7586244], США). Следует отметить, что риск развития венозных тромбозов значительно увеличивается (8-кратное увеличение), если пациент, кроме мутации Лейден фактора V, также имеет мутацию Т полиморфизма С677Т гена метилтетрагидрофолатредуктазы.
Одним из самых опасных осложнений гормональных контрацептивов являются тромбозы и тромбоэмболии. Многие женщины с такими осложнениями являются гетерозиготными носителями мутации Лейден (генотип G/A). На фоне приема гормональных контрацептивов риск тромбозов у них повышен в 6-9 раз. У женщин, использующих гормональные противозачаточные средства и имеющих гомозиготную мутацию Лейден (генотип A/A), риск развития тромбоза церебральных синусов (ТЦС) повышен более чем в 30 раз по сравнению с пациентками, не имеющих этой мутации ([9518910], Нидерланды).
Были обобщены конечные данные исследования Women’s Health Initiative Estrogen Plus Progestin о частоте венозных тромбозов на фоне заместительной гормонотерапии (ЗГТ). В исследовании приняли участие 16 608 женщин в постменопаузе в возрасте от 50 до 79 лет, наблюдавшихся с 1993 по 1998 гг. в течение 5 лет. Наличие мутации Лейден усиливало риск тромбозов при эстроген-гестагенной заместительной гормонотерапии почти в 7 раз по сравнению с женщинами без этой мутации. Присутствие других генетических мутаций (протромбин 20210А, метилентетрагидрофолатредуктаза С677Т, фактор XIII Val34Leu, PAI-1 4G/5G, фактор V HR2) не влияли на связь ЗГТ и риска венозных тромбозов.
Анализ более десяти независимых исследований показал, что среди пациентов, перенесших инфаркт миокарда до 55 лет, распространенность мутации Лейден была заметно выше. Среднестатистический риск развития инфаркта миокарда увеличивается в 1,5 раза. Более того, мутация Лейден приводит к 2,8 — кратному повышению количества пациентов без выраженного коронарного стеноза, заболевших инфарктом миокарда ([12938565], Франция).
Полиморфизм 20210 G->A протромбина
Физиология и генетика. Протромбин (коагуляционный фактор II или F2) является одним из главных компонентов системы свертываемости крови. В ходе ферментативного расщепления протромбина образуется тромбин. Данная реакция является первой стадией образования кровяных сгустков. Мутация гена протромбина G20210A характеризуется заменой нуклеотида гуанина (G) нуклеотидом аденин (A) в позиции 20210. Из-за увеличения экспрессии мутантного гена уровень протромбина может быть в полтора-два раза выше, чем в норме. Мутация наследуется по аутосомно-доминантному типу. Это означает, что тромбофилия возникает даже у гетерозиготного носителя измененного гена (G/A).
Тромбоэмболические заболевания (ТЭ) вызываются нарушениями в системе свертываемости крови. Эти нарушения приводят и к сердечно-сосудистым заболеваниям. Генотип G/A является показателем риска развития тромбозов и инфаркта миокарда. При возникновении тромбозов мутация 20210A часто встречается в сочетании с мутацией Лейден. Генотип G/A позиции 20210 гена протромбина является фактором риска тех же осложнений, которые связанны с мутацией Лейден.Гетерозиготными носителями гена являются 2-3% представителей европейской расы.
Показания к анализу. Инфаркт миокарда, повышенный уровень протромбина крови, тромбоэмболические заболевания в анамнезе, преклонный возраст пациента, невынашивание беременности, фетоплацентарная недостаточность, внутриутробная гибель плода, токсикоз, задержка развития плода, отслойка плаценты, пациентам, готовящимся к большим полостным операциям (миома матки, кесарево сечение, кисты яичников и пр.), курение.
Клинические данные. В исследовании 500 пациентов с инфарктом миокарда и 500 здоровых доноров показано более чем пятикратное увеличение риска инфаркта миокарда у пациентов с генотипом 20210A моложе 51 года ([12480694], Канада). Генетический анализ группы пациенток с первым инфарктом миокарда (возраст 18-44 лет) показал, что вариант 20210А встречается в четыре раза чаще в сравнении с группой здоровых, что соответствует увеличению риска инфаркта в 4 раза. Вероятность инфаркта была особенно высока при наличии других факторов риска сердечно-сосудистых заболеваний. Например, курение при наличии генотипа 20210А повышает риск инфаркта миокарда более чем в 40 раз ([9292507], Нидерланды). Мутация 20210А является значительным фактором риска раннего инфаркта миокарда. При исследовании пациентов с семейным анамнезом венозного тромбоза и контрольной группы здоровых доноров обнаружено, что мутация 20210A приводит к трехкратному увеличению риска венозного тромбоза. Риск тромбоза увеличивается для всех возрастов и для обоих полов. В этом исследовании также была подтверждена прямая связь между наличием мутации 20210A и повышенным уровнем протромбина в крови ([8916933], Нидерланды).
В терапевтических стационарах, где преобладают больные с сердечно-сосудистыми заболеваниями, ТЭ в форме тромбоэмболии легочной артерии встречается в 15-30% случаев. Во многих случаях ТЭ являются непосредственной причиной смерти, особенно у послеоперационных больных и больных раком. Установлено, что среди больных раком при наличии ТЭ смертность увеличивается в несколько раз, при этом количество ТЭ превышает среднестатистические значения. Причины роста ТЭ у больных раком, возможно, следует искать в проводимой терапии, несогласованной с генетической предрасположенностью больного. Это касается не только больных раком. Согласно патологоанатомическим отчетам, у 60% больных, умерших в больницах общего профиля, обнаруживают признаки тромбоэмболических заболеваний. Знание генотипических характеристик пациента позволит не только оценить риск развития угрожающих жизни состояний, но и правильно определить способы их профилактики и лечения, а также возможность применения тех или иных лекарственных препаратов.
Термолабильный вариант A222V (677 С->Т) метилентетрагидрофолатредуктазы
Клинические данные. Дефекты в данном гене часто приводят к различным заболеваниям с широким спектром клинических симптомов: умственное и физическое отставание в развитии, пренатальная смерть или дефект плода, кардиоваскулярные и нейродегенеративные заболевания, диабет, рак и другие. У носителей гетерозигот С/Т во время беременности наблюдается дефицит фолиевой кислоты, что может приводить к дефектам развития нервной трубки у плода. Курение усиливает влияние мутации. У носителей двух аллелей Т/Т (гомозиготное состояние) особенно высок риск развития побочных эффектов при приеме лекарственных препаратов, используемых в химиотерапии рака.
Гипергомоцистеинемия (ГГ) является независимым фактором риска атеросклероза и атеротромбоза (независимым от гиперлипидемии, гипертензии, сахарного диабета и т.д.). Установлено, что 10% риска развития коронарного атеросклероза обусловлено повышением уровня гомоцистеина в плазме крови. При исследовании группы пациентов с ГГ и группы здоровых доноров гомозиготная форма 677T была найдена у 73% пациентов с ГГ и только у 10% здоровых доноров. Наличие гомозиготной формы 677T приводит к почти 10-кратному повышению риска ГГ. Пациенты с ГГ также имели пониженные уровни фолиевой кислоты и витамина В12, потребляли больше кофе и курили чаще, чем здоровые доноры ([8903338], Норвегия). В норме уровень гомоцистеина равен 5-15 мкмоль/л, умеренно-повышенный уровень 15-30 мкмоль/л. При тяжелой форме ГГ возможно 40-кратное повышение уровня гомоцистеина. Исследователи приписывают причину возникновения тяжелых форм ГГ и другим мутациям и факторам (гомозиготная мутация гена Cb S, самыми частыми считают I278T и G307S, хотя частота их проявления в разных странах сильно варьирует, намного реже причинами тяжелой ГГ являются Т/Т генотип MTHFR, дефицит метионинсинтетазы и нарушенная активность метионинсинтетазы из-за генетических нарушений метаболизма витамина В12 (G. J. Hankey, J. W.Eikelboom The Lancet, 1999; 354: 407-413 http://apteki.nnov.ru/docs/294/5-1-1.html).
Коррекцию ГГ можно провести поступлением кофакторов, необходимых для метаболизма гомоцистеина (фолиевая кислота, витамины В12, В1 и В6 (особенности терапии ГГ витаминами см. G. J. Hankey, J. W.Eikelboom The Lancet, 1999; 354: 407-413 http://apteki.nnov.ru/docs/296/5-1-3.html). У носителей Т/Т генотипа MTHFR при оптимальном потреблении фолата уровень гомоцистеина повышен умеренно (до 50%). Хотя известно, что при тяжелой форме ГГ комбинация 2,5 мг фолиевой кислоты, 25 мг витамина В6 и 250 мкг витамина В12 в день снижает прогрессирование атеросклероза (измерялась бляшка в сонной артерии), нужно еще подтвердить, предупреждает ли гомоцистеин-снижающая терапия значимые сосудистые осложнения у лиц с умеренной ГГ. О важности проблемы ГГ говорит тот факт, что Министерство здравоохранения США в 1992 году рекомендовало женщинам, которые могут забеременеть, принимать 400 мкг фолиевой кислоты в день. Администрация по контролю пищевых продуктов и лекарственных препаратов в США требует обогащать крупы фолиевой кислотой в концентрациях, которые могут дать дополнительно 100 мкг в день. Однако, дневная доза фолиевой кислоты, необходимая для максимального снижения уровня гомоцистеина, равна 400 мкг, то есть могут быть оправданы и более высокие дозы добавок фолиевой кислоты в пищу.
Патогенез врожденных дефектов нервной трубки включает в себя, в частности, генетические и диетические факторы. При исследовании 40 детей Южной Италии с врожденным дефектом нервной трубки и здоровых доноров было показано, что генотип 677С в гомозиготном состоянии (С/С) приводит к двукратному повышению риска развития дефектов, в то время как мутантная гомозигота Т/Т соответствует почти десятикратному снижению риска ([12812837], Франция-Италия). При исследовании выборки населения Ирландии (395 больных и 848 здоровых) было установлено, что встречаемость варианта T повышена у пациентов с врожденным дефектом нервной трубки ([15155469], Ирландия). Трудно сказать связаны ли эти противоположные результаты исследований с популяционными изменениями или не учтены другие факторы риска. Поэтому пока нельзя определить является ли вариант Т защитным или, наоборот, патогенным фактором для данного заболевания. Повышение частоты генотипа 677T было отмечено не только при позднем токсикозе (гестозе), но и при других осложнениях беременности (отслойке плаценты, задержке роста плода, пренатальной смерти плода). Сочетание мутации 677T с другими факторами риска приводит к повышению вероятности раннего выкидыша.
При исследовании связи между мутацией 677T и сердечно-сосудистыми заболеваниями обнаружено, что гомозиготная мутация 677T встречается гораздо чаще у пациентов с кардиоваскулярными заболеваниями, чем у здоровых доноров. У молодых пациентов, имевших ишемию артерий, гомозигота Т/Т встречается в 1,2 раза чаще ([14660985], США). Статистический анализ 40 независимых исследований (мета-анализ) пациентов с ИБС, обобщающий данные о 11162 пациентах и 12758 здоровых доноров, показал увеличение риска развития ИБС в 1,16 раза при наличии гомозиготы Т/Т ([12387655], Нидерланды). Невысокая степень риска связана с гетерогенностью анализируемых выборок населения. При исследовании гомогенных выборок населения (индивидуальные исследования, а не мета-анализ) оценки степени риска значительно выше. Так, разница в частотах встречаемости гомозигот Т/Т у пациентов и у здоровых доноров соответствовала 3-х кратному повышению риска кардиоваскулярных заболеваний в раннем возрасте ([8554066], Нидерланды).Наличие мутации 677Т в гене MTHFR у больных с антифосфолипидным синдромом коррелирует с рецидивирующим течением тромбозов (Россия).
Выявлена определенная, хотя и сложная, взаимосвязь между вариантами MTHFR и развитием предраковых и раковых состояний колоректальной области. Проводилось исследование значительной группы больных с полипозом толстого кишечника. Определялись уровни фолата в эритроцитах, наряду с оценкой С/Т генотипа МТHFR. Ранее полученные результаты показывали связь между пониженным содержанием фолата и риском развития аденоматоза. Многофакторный анализ показал, что курение, фолатный статус и генотип MTHFR являются существенными компонентами высокого риска аденоматоза. Этот риск оказался весьма велик у лиц с низким уровнем фолата и носительством аллеля 677Т в гомо- или гетерозиготной форме. Эти данные показали сильное взаимодействие диетических и генных факторов в развитии предраковых состояний. Сходные предположения выдвинуты учеными, которые обследовали большой контингент больных раком толстого кишечника и показали значительную связь между риском развития ракового заболевания, возрастом больных, возрастным дефицитом фолата и Т/Т генотипом MTHFR. Исследование 379 пациентов с колоректальной аденомой и 726 здоровых доноров показало, что мужчины-носители Т/T генотипа, употребляющие много алкоголя, имели в 3,5 раза более высокий риск заболевания аденомой ([14578131], США). Однако некоторые исследователи считают, что без употребления алкоголя, как одного из факторов риска, мутация 677T является защитным фактором. Так, исследование пациентов с проксимальным колоректальным раком показало, что наличие у пациента гомозиготы Т/T приводит к 2,8-кратному понижению риска развития колоректального рака ([12576444], Италия). Эти выводы требуют проверки для других популяций. Скорее всего, значимость малоактивного мутантного MTHFR можно считать усугубляющей на фоне остальных перечисленных факторов риска, поскольку этот генный дефект может снижать стабильность генома из-за гипометилирования ДНК.
Полиморфизм С677T влияет на эффективность химиотерапии рака. Фторурацил широко используется для химиотерапии колоректального рака. Вероятность положительной динамики в ответ на химиотерапию колоректальной аденокарциномы при наличии у пациента 677T генотипа увеличивалась почти в три раза. Результаты позволяют предположить, что генотипирование по полиморфизму С677T позволит разработать более эффективные курсы химиотерапии ([12738713], Канада). Однако исследование небольших выборок (до 50) больных раком груди показало, что при наличии гомозиготы Т/Т риск развития побочных эффектов при применении метотрексата (антиметаболита, действие которого связано с ингибированием активности фермента MTHFR) увеличивается в десятки раз ([12471611], Италия).
Имеются немногочисленные исследования генотипа MTHFR при онкогинекологических заболеваниях. Изучался полиморфизм С677Т гена MTHFR в большой группе еврейских женщин, заболевших раком молочной железы и яичника, включая и наследственные формы, связанные с мутациями генов BRCA. При таком неблагоприятном генетическом фоне наличие у больных Т/Т генотипа оказалось существенным фактором отягощения заболевания. Частота Т/Т генотипа была в 2 раза выше (33% против 17%, Р=0,0026) среди женщин с двусторонним раком молочной железы и раком яичника, по сравнению с основной группой больных. Женщины с гетерозиготным генотипом С/Т имели двойной онкологический риск, а у больных с гомозиготным генотипом Т/Т риск был повышен втрое по сравнению с контрольной группой. В то же время, пониженное потребление фолатов в диете повышало генетический риск до пятикратного значения по сравнению с контролем. Авторы также подтвердили тот факт, что заражение HPV (вирус папилломы) у больных является важнейшим фактором риска развития цервикальной дисплазии. В то же время подчеркивается особое значение сочетания HPV-инфекции с Т/Т вариантом MTHFR (Gershoni-Baruch R., Dagan E., Israeli D. et al. Association of the C677T polymorphism in the MTHFR gene with breast and/or ovarian cancer risk in Jewish women // Eur. J. Cancer. — 2000. — Vol.36, N.18. — P. 2313-2316).
Полиморфизм Arg353Gln (10976 G->A) коагуляционного фактора VII (F7)
Физиология и генетика. В активном состоянии фактор VII взаимодействует с фактором III, что приводит к активации факторов IX и X системы свертывания крови, то есть коагуляционный фактор VII участвует в образовании кровяного сгустка. Вариант 353Gln (10976A) приводит к понижению производительности (экспрессии) гена фактора VII и является защитным фактором в развитии тромбозов и инфаркта миокарда. Распространенность данного варианта в европейских популяциях составляет 10-20%.
Показания к анализу. Риск инфаркта миокарда и фатального исхода при инфаркте миокарда, уровень коагуляционного фактора VII в крови, тромбоэмболические заболевания в анамнезе.
Клинические данные. Высокий уровень коагуляционного фактора VII в крови связывают с повышенным риском смерти при инфаркте миокарда [Meade TW et al, Lancet 1986, 2: 533-7]. Приведенные данные о клинической значимости мутации подтверждаются исследованиями в других европейских популяциях. В частности, наличие варианта 10976A соответствовало пониженному риску фатального исхода при инфаркте миокарда ([11858502], Финляндия). При исследовании пациентов со стенозом коронарных артерий и инфарктом миокарда обнаружено, что наличие мутации 10976A приводит к понижению уровня фактора VII в крови на 30% и 2-х кратному понижению риска инфаркта миокарда даже при наличии заметного коронарного атеросклероза. В группе пациентов, не имевших инфаркта миокарда, наблюдалась повышенная встречаемость гетеро- и гомозиготных генотипов 10976A, соответственно G/A и G/G ([10984565], Италия).
Полиморфизм L33P (T->C) тромбоцитарного рецептора фибриногена, гликопротеина-3а
Физиология и генетика. Ген тромбоцитарного рецептора фибриногена (ITGB3) кодирует бета-3 субъединицу интегрин-комплекса поверхностного рецептора тромбоцитов GPIIb/IIIa, известную также как гликопротеин-3а (GPIIIa). ITGB3 участвует в межклеточной адгезии и сигнализации. ITGB3 обеспечивает взаимодействие тромбоцита с фибриногеном плазмы крови, что приводит к быстрой агрегации (склеиванию) тромбоцитов и, таким образом, к последующему купированию поврежденной поверхности эпителия. Мутация 33P GPIIIa приводит к повышенной склонности тромбоцитов к агрегации, что увеличивает риск развития сердечно-сосудистых заболеваний. У пациентов с этим вариантом часто отмечается пониженная эффективность аспирина как тромболитического препарата.Частота встречаемости мутации 33P в европейских популяциях составляет 8-15%.
Показания к анализу. Семейный анамнез ранней ишемической (коронарной) болезни сердца (ИБС), инфаркт миокарда, тромбоэмболические заболевания в анамнезе, постангиопластические тромбозы, неонатальная тромбоцитопения, антитромбозная терапия аспирином.
Клинические данные. Исследование европейской популяции и афро-американских близнецов-пациентов с коронарной болезнью сердца показывает повышенную встречаемость мутации 33P в семьях с ранней ИБС. Наличие генотипа 33P приводит к увеличению риска заболевания инфарктом миокарда в 2,8 раза. Для пациентов с генотипом 33P моложе 60 лет среднестатистический риск увеличивается более чем в 6 раз ([10583927], [8598867], США).
Аспирин широко применяется в антикоагуляционной терапии с целью понижения риска развития инфаркта миокарда. Многочисленные исследования, включающие более 100000 пациентов, показывают значительное уменьшение среднестатистической смертности, инфаркта и окклюзии артерий при применении аспирина [8054013]. Полиморфизм L33P может определять реакцию организма пациента на терапию аспирином. Обнаружено, что в группе пациентов, не реагирующих на прием аспирина, частота встречаемости немутантного ITGB3 (вариант 33L) была почти в 1,5 раза выше, чем в контрольной группе. Иначе говоря, мутантный GPIIIa (вариант 33P) способствует повышению чувствительности к терапии аспирином. Таким образом, генетическое тестирование полиморфизма L33P гена GPIIIa может быть использовано для выработки индивидуального курса терапии аспирином ([13678940], Франция).
Полиморфизм –455 G->A фибриногена
Физиология и генетика. При повреждении кровеносных сосудов фибриноген переходит в фибрин — основной компонент кровяных сгустков (тромбов). Мутация -455А бета фибриногена (FGB) сопровождается повышенной производительностью (экспрессией) гена, что приводит к повышенному уровню фибриногена в крови и увеличивает вероятность образования тромбов. Распространенность данного варианта в европейских популяциях составляет 5-10%.
Показания к анализу. Повышенный уровень фибриногена плазмы крови, повышенное давление крови, тромбоэмболические заболевания в анамнезе, инсульт.
Клинические данные. Повышенная склонность к тромбообразованию может приводить к тромбозам и кардиоваскулярным заболеваниям. Уровень фибриногена в крови определяется рядом факторов, среди которых прием лекарственных препаратов, курение, прием алкоголя и вес тела. Однако и генотипам G и A соответствует заметная разница в уровнях фибриногена крови (10-30% по различным исследованиям).
В исследовании группы здоровых доноров было установлено, что мутация -455А приводит к повышенному содержанию фибриногена в крови ([10591688], Швеция). В крупномасштабном исследовании EUROSTROKE было установлено, что риск инсульта (ишемического или геморрагического) повышается в 2-3 раза при увеличении содержания фибриногена крови. Риск дополнительно увеличивается при повышенном систолическом давлении (>160 мм рт. ст.) ([11815639], Нидерланды). Эти данные подтверждаются исследованиями неевропейских популяций. При повышенном давлении крови наличие генотипа -455А повышает риск развития ишемического инсульта ([9690809], Япония). Пациенты с инсультом, имеющие генотип -455А, характеризуются многоочаговостью поражений: могут иметь три или более лакунарных инфаркта церебральных сосудов, в среднем риск инсульта увеличивается в 2,6 раза. При повышенном давлении крови у пациентов с мутацией риск многоочагового инсульта повышается более чем в 4 раза ([12637691], Финляндия).
Алкогольная зависимость
Полиморфизмы генов метаболизма алкоголя
Полиморфизмы Rsal (-1019 C->T), Pstl (-1293 G->C), Dral (T->C) и Tag 1 (9896 C->G) цитохрома 2E1
Физиология и генетика. Цитохром 2E1 (CYP2E1) участвует в метаболизме ацетона, бензола, бензопирена, тетрахлористого углерода и других канцерогенов табачного дыма. Фермент также участвует в метаболизме этанола. В результате ферментативных реакций цитохрома образуются перекись водорода и свободнорадикальные пероксид и гидроксил (из этанола), что вызывает повреждения органов и, прежде всего печени. Следовательно, изменение количества или активности фермента приводит к изменению риска повреждения организма.
Вариант T RsaI (Rsal c2) ассоциирован с алкогольной болезнью печени. Вариант C PstI (PstI+) соответвует повышенному риску развития онкологических заболеваний. Вариант C DraI также является онкологическим маркером.В европейских популяциях частоты встречаемости вариантов RsaI и PstI составляют 1-3%, варианта DraI — около 10%.
Показания к анализу. Прием алкоголя, алкоголизм, алкогольное повреждение печени, гепатома (форма первичного рака печени), колоректальный рак, рак желудка, рак груди, рак простаты, курение.
Клинические данные. Полиморфизм Rsal (-1019 C->T). При исследовании больных алкоголизмом и группы здоровых доноров было установлено, что частота встречаемости варианта RsaI-Т была в 4,5 раза выше в группе больных со значительными повреждениями печени ([8747406], Англия). Исследование пациентов с гепатомой печени и здоровых доноров показало, что частоты встречаемости вариантов RsaI-С/Т незначительно отличаются между двумя группами. Однако, у пациентов с RsaI-Т, употреблявших алкоголь (более 50 г в день), риск развития гепатомы повышается почти в 5 раз ([8977352], Испания).
Полиморфизм Pstl (-1293 G->C). Исследование групп больных с колоректальным раком показало увеличение риска заболевания при наличии PstI-C в 1,9 раза. Выявлено, что при наличии PstI-C в 3 раза увеличивается риск развития рака в подгруппе пациентов, регулярно употребляющих соленую пищу ([15182482], Китай). Многофакторный анализ по возрасту и количеству выкуриваемых сигарет группы пациентов с раком желудка и здоровых доноров без онкологической предыстории показал, что частоты встречаемости полиморфизма PstI значительно различаются между группами. Так, наличие PstI-C при выкуривании более 30 пачек сигарет в год приводит к 3-х кратному увеличению риска развития рака желудка. У пациентов моложе 50 лет при наличии гомозиготы PstI-C/С или DraI-C/С риск развития рака повышался в 5,6 раза ([14535982], Корея).
Полиморфизм Dral (T->C). Известно, что N-нитрозамины табачного дыма канцерогенны и вызывают, в частности, рак груди. Нитрозамины также выводятся CYP2E1. В США проводилось исследование пациенток до и после менопаузы. Было показано, что наличие DraI-C у курящих пациенток до менопаузы является фактором риска заболевания раком груди. Даже при незначительном курении (1 сигарета в неделю) более одного года наличие у пациенток гетерозиготного генотипа DraI-Т/С приводит к увеличению риска заболевания раком груди более чем в 7 раз ([8944074], США).
Полиморфизм ADh2B*2 (R47H G->A) алкогольдегидрогеназы
Физиология и генетика. Алкогольдегидрогеназа 1B (ADh2B, ADh3) окисляет спирты до альдегидов. Мутация 47H наиболее распространенного полиморфизма ADh2B*2 приводит к повышенной скорости распада этанола, тем самым, ускоряя удаление спирта из крови. Это благоприятная мутация для употребляющих алкоголь. Распространенность мутации составляет 4-8% в европейских популяциях.
Показания к анализу. Алкоголизм, алкогольный цирроз печени, риск инсульта, рак груди.
Клинические данные. Исследование большой выборки населения (876 человек) из различных европейских стран (Испания, Франция, Германия, Швеция, Польша) показало, что встречаемость варианта ADh2B*2 гораздо ниже у больных алкоголизмом. Риск алкоголизма при наличии варианта ADh2B*2 уменьшается на 30% ([10733556], Испания). Аналогичные результаты были получены в отношении ряда неевропейских выборок населения (например, [14745297]).
В исследовании пациентов с циррозом не было обнаружено мутации ADh2B*2, в то время как она была найдена, по крайней мере, у 8% здоровых доноров. Сделан вывод, что наличие варианта ADh2B*2 соответствует значительному понижению риска алкогольного цирроза печени ([11966948], Австралия). При исследовании более 2000 человек было установлено, что мутация ADh2B*2 является защитным фактором, понижающим риск инсультов ([15534263], Япония). Исследование 278 пациенток с инвазивным раком груди и 467 здоровых доноров показало в три раза более высокую частоту варианта ADh2B*2 у пациенток с раком груди ([12189549], Германия).
Полиморфизм ALDh3*2 (E487K G->A) альдегиддегидрогеназы
Физиология и генетика. Альдегиддегидрогеназа (ALDh3) принимает участие в распаде спиртов — переводит образующиеся после окисления спиртов альдегиды в кислоты. Большинство европейцев имеют два основных изофермента: цитозольный и митохондриальный, в то время как почти половина исследованных представителей азиатских популяций имеют только цитозольную форму. Ген ALDh3 кодирует митохондриальную форму фермента. Отсутствие его активности является, по-видимому, причиной более сильного воздействия алкоголя, наблюдаемого у азиатских и северных народностей.Полиморфизм ALDh3*2 встречается у 50% представителей монголоидной расы и практически не встречается у представителей кавказской и негроидной рас.
Показания к анализу. Принадлежность к монголоидной расе, алкоголизм, алкогольный цирроз печени, повышенный уровень ЛПВП-холестерина, инфаркт миокарда, периодонтит.
Клинические данные. У пациентов с мутацией ALDh3*2 происходит накопление ацетальдегида в крови, приводящее к поражению печени. Интенсивный прием алкоголя у лиц с указанным полиморфизмом может привести, в частности, к быстрому развитию алкогольной болезни и/или циррозу печени. Исследование пациентов, находящихся на различных стадиях алкогольного повреждения печени показало, что гетерозиготный генотип G/A ALDh3*2 приводит к более серьезному повреждению печени. В частности, у пациентов с мутацией ALDh3*2, регулярно принимавших небольшие количества алкоголя, повреждения печени были сильнее, чем у пациентов без этой мутации, принимавших более 100 г этанола в день ([12198368], Япония). При исследовании 342 пациентов-мужчин с инфарктом миокарда и 1820 здоровых доноров была показана связь варианта 487K с количеством потребления алкоголя и уровнем ЛПВП-холестерина. При наличии варианта 487K риск инфаркта миокарда повышается в 1,6 раз ([12452318], Япония) Исследование пациентов с различной степенью периодонтита показало, что прием алкоголя увеличивает риск развития периодонтита в 2 раза вне зависимости от варианта E487K. Наличие же варианта 487K соответствовало более тяжелым формам периодонтита при приеме более 30 г этанола в день ([14742656], Япония).
Полиморфизмы генов системы детоксикации организма человека
Полиморфизмы R144C (C->T) и I359L (A->C) цитохрома 2C9
Физиология и генетика. Цитохром 2C9 (CYP2C9) является одним из ферментов системы детоксикации организма от ксенобиотиков, влияет на эффективность воздействия ряда лекарственных препаратов. Варианты 144C (CYP2C9*2) и 359L (CYP2C9*3) определяют индивидуальную чувствительность пациента к антикоагуляционной терапии варфарином, а также при применении ряда других лекарственных препаратов (аценокумарол, толбутамид, лозартан, глипизид, фенитоин, ибупрофен).Частота встречаемости варианта CYP2C9*2 составляет 11%, а варианта CYP2C9*3 – 7% в европейских популяциях.
Показания к анализу. Антикоагуляционная терапия варфарином и аценокумаролом, риск кровоизлияний, колоректальный рак.
Клинические данные. Уменьшение активности фермента генотипа 144C или 359L приводит к риску развития осложнений при фармакотерапии. При назначении курса лечения обязательно нужно принимать во внимание данные генетического анализа CYP2C9. Например, в случае лечения варфарином пациента с вариантом 144C или 359L следует назначать сниженные дозы данного препарата (см. лекцию по генотипированию на сайте www.pynny.ru в разделе Информация). Исследование группы пациентов, проходящих курс терапии варфарином, показало, что при наличии одного из генотипов 144C или 359L содержание варфарина в крови превышает принятые нормы. Кроме того, у пациентов с такими аллелями риск серьезного кровоизлияния, угрожающего жизни пациента, повышается в 2,4 раза ([11926893], США). Исследование пациентов, проходящих терапию аценокумаролом, показало, что вариант 359L встречается в 6 раз чаще у тех, которым из-за побочных клинических эффектов назначалась пониженная доза препарата. Иначе говоря, при наличии варианта 359L необходимо снижать дозы аценокумарола. При наличии варианта 144C также необходимо использовать пониженные дозы препарата. Однако у пациентов с вариантом 144C побочные клинические эффекты были гораздо менее выражены. Сделан вывод, что аценокумарол может использоваться как альтернатива варфарину при наличии у больного варианта 144C (но не варианта 359L) ([12010835], Испания).
Некоторые из субстратов CYP2C9 участвуют в метаболизме веществ, влияющих на риск развития колоректального рака. Таким образом, полиморфные варианты CYP2C9 характеризуются разным онкологическим риском. Исследование пациентов с колоректальным раком и группы здоровых доноров показало, что для больных характерна более высокая частота нативной формы фермента, в особенности для пациентов с раком проксимальных сегментов прямой кишки. Наличие хотя бы одного из вариантов 144C или 359L приводит к более чем двукратному понижению риска развития колоректального рака ([11470765], Испания).
Источник
Использованы литературные данные, подготовленные к.х.н Торшиным И.Ю., к.б.н. Генерозовым Э.В., Третьяковым В.Е.
Расширенное исследование генов системы гемостаза (без описания результатов врачом-генетиком)
Метод определения Real-time-PCR.
Исследуемый материал Цельная кровь (с ЭДТА)
Доступен выезд на дом
Расширенное исследование генов системы гемостаза: F2, F5, MTHFR, MTR, MTRR, F13, FGB, ITGA2, ITGВ3, F7, PAI-1
Комплексное исследование генетических факторов риска развития нарушений в системе свертывания крови и фолатном цикле (без заключения врача-генетика).
Различные изменения в генах системы гемостаза и цикла обмена фолатов предрасполагают к развитию большого числа патологических состояний: инфаркты, инсульты, тромбоэмболии, кровотечения, патология беременности и родов, осложнения послеоперационного периода и т.д. Профиль включает в себя исследование основных полиморфизмов в генах системы гемостаза и фолатного цикла:- F2 c.*97G>A (20210 G>A; rs1799963),
- F5 c.1601G>A (Arg534Gln; 1691 G>A; rs6025),
- MTHFR c.665C>T (Ala222Val; 677 C>T; rs1801133),
- MTHFR c.1286A>C (Glu429Ala; 1298 A>C; rs1801131),
- MTR c.2756A>G (Asp919Gly; rs1805087),
- MTRR c.66A>G (Ile22Met; rs1801394),
- F13 с.103G>T (I63Т; rs5985),
- FGB c.-467G>A (-455 G>А; rs1800790),
- ITGA2 c.759C>T (Phe253Phe, 807 C>T; rs1126643),
- ITGB3 c.176T>C (Leu59Pro; 1565 T>C; rs5918),
- F7 c.1238G>A (Arg353Gln; 10976 G>A; rs6046),
- PAI-1 (SERPINE1) –675 5G>4G (rs1799889).
*Примечание: иногда в научной литературе при описании однонуклеотидных замен, характерных для генных полиморфизмов, встречается термин "мутантный аллель". Это терминологическая неточность, так как в классической генетике термин "мутантный аллель" традиционно рассматривается как синоним термина "мутация". При мутациях, как известно, изменение структуры гена приводит к образованию (экспрессии) нефункциональных белков и к неизбежному развитию наследственного заболевания. При полиморфизмах изменение в структуре гена приводит лишь к появлению белков с немного изменёнными физико-химическими свойствами. Такие изменения, как известно, проявляют себя при воздействии на организм различных факторов внешней среды или при изменении функционального состояния организма человека. И только в таких ситуациях функционирование белков со структурными особенностями может, либо способствовать ускорению развития заболевания, либо, напротив, тормозить формирование патологических процессов. Поэтому, на наш взгляд, для разграничения изменений в генах столь очень похожих структурно, но приводящих к несоизмеримо разным последствиям для организма, корректнее в отношении генных полиморфизмов применять понятие "аллельный вариант гена", а не "мутантный аллель".
Литература
- Никитина Л.А. и др. Роль некоторых генетических полиморфизмов в невынашивании беременности // Проблемы репродукции, 2007, С.83-89.
- Güngör et al. The presence of PAI-1 4G/5G and ACE DD genotypes increases the risk of early-stage AVF thrombosis in hemodialysis patients. // Ren Fail. 2011;33(2):169-7
- Wei YS, et al. Association of the integrin gene polymorphisms with ischemic stroke and plasma lipid levels // Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2009;26(2):211-5
- Gohil et al., The genetics of venous thromboembolism. A meta-analysis involving approximately 120,000 cases and 180,000 controls // Thromb Haemost, 2009. 102(2): 360-70
- Goodman et al., Which thrombophilic gene mutations are risk factors for recurrent pregnancy loss? // Am J Reprod Immunol, 2006. 56(4):230-6
- Gerhardt, A., et al. The polymorphism of platelet membrane integrin alpha2beta1 (alpha2807TT) is associated with premature onset of fetal loss // Thromb Haemost, 2005. 93(1):124-9.
- Ruzzi, L., et al., Association of PLA2 polymorphism of the ITGB3 gene with early fetal loss // Fertil Steril, 2005. 83(2): 511-2
- База OMIM: http://omim.org/entry/176930
- База OMIM: http://omim.org/entry/227400
- База OMIM +227400 http://omim.org/entry/607093
- База OMIM: http://omim.org/entry/602568
- База OMIM: http://omim.org/entry/156570
- http://www.ncbi.nlm.nih.gov/clinvar/RCV000012861/
Однонуклеотидный полиморфизм — Википедия
В случае SNP гетерогенность первичной структуры ДНК проявляется в однонуклеотидных различиях аллелейОднонуклеотидный полиморфизм (ОНП, англ. Single nucleotide polymorphism, SNP, произносится как снип) — отличия последовательности ДНК размером в один нуклеотид (A, T, G или C) в геноме (или в другой сравниваемой последовательности) представителей одного вида или между гомологичными участками гомологичных хромосом.
Если две последовательности ДНК — AAGCCTA и AAGCTTA — отличаются на один нуклеотид, в таком случае говорят о существовании двух аллелей: C и T. Однонуклеотидные полиморфизмы (SNPs) возникают в результате точечных мутаций.
Однонуклеотидный полиморфизм (наряду с полиморфизмом длин рестрикционных фрагментов (англ. RFLP) и ПДАФ (англ. AFLP)) широко используют в качестве молекулярно-генетических меток (ма́ркеров) для построения кладограмм молекулярно-генетической систематики на основе дивергенции (расхождения) гомологичных участков ДНК в филогенезе. В данной области наиболее часто используются спейсеры генов рибосомальной РНК. Ввиду того, что мутации в данных спейсерах не сказываются на структуре конечных продуктов гена (теоретически они не влияют на жизнеспособность), в первом приближении постулируется прямая зависимость между степенью полиморфизма и филогенетическим расстоянием между организмами.
Единой номенклатуры для SNPs нет: часто существуют несколько различных вариантов названия для одного конкретно выбранного SNP, к какому-то согласию в этом вопросе прийти пока не удается. Один из подходов – писать SNPs с префиксом, точкой и знаком «больше чем», показывающим нуклеотид или аминокислоту дикого типа и измененную (например, c.76A>T).[1]
Однонуклеотидный полиморфизм встречается в пределах кодирующих последовательностей генов, в некодирующих участках или в участках между генами. SNPs, встречающиеся в кодирующих участках, могут не менять аминокислотную последовательность белка из-за вырожденности генетического кода.
Однонуклеотидные полиморфизмы кодирующих участков бывают двух типов: синонимические и несинонимические. Синонимические SNPs оставляют аминокислотную последовательность белка без изменения, тогда как несинонимические SNPs изменяют её. Несинонимические SNPs можно разделить на missense и nonsense. Однонуклеотидный полиморфизм, встречающийся в некодирующих участках гена, возможно, влияет на генетический сплайсинг, деградацию мРНК, связывание транскрипционных факторов.
Разнообразием последовательностей ДНК у людей, возможно, объясняется то, как у них происходит течение различных заболеваний, реакции в ответ на патогены, прием лекарств, вакцин и т.п. Огромное значение SNPs в биомедицинских исследованиях состоит в том, что их используют для сравнения участков генома между исследуемыми группами (например, одна группа – люди с определенным заболеванием, а вторая – без него).[5]
Однонуклеотидные полиморфизмы также используют в GWAS в генетическом картировании как маркеры с высоким разрешением, благодаря их количеству и стабильной наследуемости в ряду поколений. Знание об однонуклеотидном полиморфизме, вероятно, поможет в понимании фармакокинетики и фармакодинамики действия различных лекарств на человека. Широкий спектр заболеваний, такие как рак, инфекционные аутоиммунные заболевания, серповидноклеточная анемия и многие другие, возможно, возникают из-за однонуклеотидного полиморфизма.[6]
Для SNPs существует большое количество баз данных. Ниже приведены некоторые из них.
dbSNP[7] – база данных SNP, свободный общественный архив, содержащий данные по наследственной изменчивости различных видов, разработанный и поддерживаемый NCBI (National Center for Biotechnology Information Национальный центр биотехнологической информации США). Хотя такое название базы данных подразумевает, что там собран только один класс полиморфизмов, а именно SNPs, на самом деле она содержит большое количество информации и о других молекулярных изменениях в аминокислотных последовательностях. dbSNP была создана в сентябре 1998 года в дополнение к GenBank, в котором представлены нуклеотидные и аминокислотные последовательности, находящиеся в свободном доступе.[8] К 2010 году dbSNP содержала больше 184 миллионов последовательностей, представляя более 64 миллионов различных вариантов для 55 организмов, включая Homo sapiens, Mus musculus, Oryza sativa и множество других. Полный список организмов можно найти по ссылке: https://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi
SNPedia – биоинформатический вики-сайт, который служит как база данных SNP. Каждая статья про SNPs предоставляет краткое описание, ссылки на научные статьи, и, кроме того информацию с микрочипа про однонуклеотидный полиморфизм данного типа. SNPedia помогает в интерпретации результатов собственной генетической информации с помощью таких программ как, например, Promethease, 23andMe, Navigenics, deCODEme или Knome.[9] SNPedia была создана и поддерживается генетиком Грегом Ленноном и программистом Майком Кариазо. К 14 сентября 2017 года в базе данных содержалось 107 125 однонуклеотидных полиморфизмов.[10]
База данных GWAS Central выдает краткое содержание объединенных данных в одном или нескольких полно-геномных исследованиях. В этой базе данных представлено наиболее полное собрание p-value ассоциаций. GWAS Central использует мощные графические и текстовые методы представления данных для открытия и одновременной визуализации многих однонуклеотидных полиморфизмов. Исследователям также предоставляется возможность просматривать их персональные данные рядом с выбранными. Кроме того, данные находятся в свободном доступе для скачивания их научными сообществами.
International HapMap Project – организация, целью которой является развитие карты гаплотипов человеческого генома, которая будет описывать общие паттерны генетической изменчивости у людей. HapMap – основной ресурс для выявления генетической изменчивости, влияющую на здоровье, факторы окружающей среды и т.п. Вся предоставляемая информация находится в свободном доступе. Этот проект – результат сотрудничества различных групп ученых из Канады, Китая, Японии, Нигерии, Великобритании и США, окончательная версия которого увидела свет весной 2009 года. В определенном участке генома располагается набор свободных SNP (Tag SNP), которые хорошо коррелируют со всеми остальными SNP в данном участке. Далее, изучив аллели свободных SNP, можно с большей вероятностью определить гаплотип индивидуума. Так определяют гаплотипы у нескольких представителей (некоторые болеют определенным заболеванием, а другие нет), а потом, сравнивая две группы, определяют наиболее вероятное расположение SNP и гаплотипов, которые вовлечены в заболевание.
MirSNP – база данных однонуклеотидных полиморфизмов, изменяющих сайты связывания микроРНК. В ней содержится 12 846 SNPs, включая 1940 SNPs в пре-микроРНК.[11]
Аналитические методы открытия новых SNP и обнаружения уже известных SNPs включают:
1. Гибридизационные методы
- Принцип молекулярных маяков (англ. Molecular Beacons)
Суть этого принципа в том, что концы пробы (на которых находятся соответственно метка и тушитель флуоресценции) комплементарны друг другу. В результате при температуре отжига праймеров они схлопываются и образуют структуру типа "ручки сковородки" (stem-loop), где зона комплементарности пробы с матрицей находится в петле. При гибридизации пробы с матрицей вторичная структура разрушается, флуоресцентная метка и тушитель расходятся в разные стороны, и флуоресценция от метки может быть детектирована.
2. Ферментативные методы
3. Методы, основанные на физических свойствах ДНК:
4. секвенирование ДНК[14]. Для картирования SNP на протяжении всего генома сейчас применяют методы секвенирования нового поколения.
- ↑ J.T. Den Dunnen. Recommendations for the description of sequence variants (англ.) // Human Genome Variation Society : journal. — 2008.
- ↑ Giegling I., Hartmann A.M., Möller H.J., Rujescu D. Anger- and aggression-related traits are associated with polymorphisms in the 5-HT-2A gene (англ.) // Journal of Affective Disorders (англ.)русск. : journal. — 2006. — November (vol. 96, no. 1—2). — P. 75—81. — doi:10.1016/j.jad.2006.05.016. — PMID 16814396.
- ↑ Morita, Akihiko; Nakayama, Tomohiro; Doba, Nobutaka; Hinohara, Shigeaki; Mizutani, Tomohiko; Soma, Masayoshi. Genotyping of triallelic SNPs using TaqMan PCR (англ.) // Molecular and Cellular Probes (англ.)русск. : journal. — 2007. — Vol. 21, no. 3. — P. 171—176. — doi:10.1016/j.mcp.2006.10.005. — PMID 17161935.
- ↑ Ammitzbøll, Christian Gytz; Kjær, Troels Rønn; Steffensen, Rudi; Stengaard-Pedersen, Kristian; Nielsen, Hans Jørgen; Thiel, Steffen; Bøgsted, Martin; Jensenius, Jens Christian. Non-Synonymous Polymorphisms in the FCN1 Gene Determine Ligand-Binding Ability and Serum Levels of M-Ficolin (англ.) // PLoS ONE : journal. — 2012. — 28 November (vol. 7, no. 11). — P. e50585. — doi:10.1371/journal.pone.0050585.
- ↑ Carlson et al. SNPs — A Shortcut to Personalized Medicine (неопр.) // Genetic Engineering & Biotechnology News. — 2008.
- ↑ Ingram et al. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin (англ.) // Nature : journal. — 1956.
- ↑ Wheeler et al. Database resources of the National Center for Biotechnology Information (англ.) // Nucleic Acids Res (англ.)русск. : journal. — 2007.
- ↑ Sherry et al. dbSNP - database for single nucleotide polymorphisms and other classes of minor genetic variation (англ.) // Genome Research (англ.)русск. : journal. — 1999.
- ↑ Michael Cariaso. SNPedia: A Wiki for Personal Genomics (неопр.) // Bio-IT World. — 2007.
- ↑ Michael Cariaso and Greg Lennon. SNPedia: a wiki supporting personal genome annotation, interpretation and analysis (англ.) // Nucleic Acids Research (англ.)русск. : journal. — 2011.
- ↑ Chenxing Liu et al. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs (англ.) // BMC Genomics (англ.)русск. : journal. — 2012.
- ↑ Drabovich et al. Identification of base pairs in single-nucleotide polymorphisms by MutS protein-mediated capillary electrophoresis (англ.) // Analytical chemistry : journal. — 2006.
- ↑ Griffin et al. Genetic identification by mass spectrometric analysis of single-nucleotide polymorphisms: ternary encoding of genotypes (англ.) // Analytical chemistry : journal. — 2000.
- ↑ Altshuler et al. An SNP map of the human genome generated by reduced representation shotgun sequencing (англ.) // Nature : journal. — 2000.
Анализ Фолатный цикл, полиморфизм генов (MTHFR, MTR, MTRR)
Диагностическое направление
Оценка системы гемостаза
Общая характеристика
Фолаты – производные фолиевой кислоты (витамина B9), играющие ведущую роль в широком спектре жизненно важных процессов: стимулируют эритропоэз, участвуют в синтезе аминокислот, нуклеиновых кислот, пуринов, пиримидинов, витаминов, участвуют в обмене холина, гистидина, в метилировании ДНК и РНК, способствуют регенерации мышечной ткани, развитию быстро растущих тканей (кожа, оболочки ЖКТ, костный мозг), выполняют защитную роль при беременности от тератогенных и повреждающих факторов на плод, способствуют созреванию и функционированию плаценты, оказывают эстрогеноподобное действие. Данные функции реализуются в процессе Фолатного цикла – это каскадный процесс синтеза аминокислоты метионина из гомоцистеина, контролируемый 3-мя ферментами: метилентетрагидрофолатредуктазой (MTHFR), метионин-синтазой (MTR) и метионин-синтаза-редуктазой (MTRR). В фолатном цикле фолаты являются коферментами. Главные причины нарушения фолатного цикла: 1. Генетические дефекты ферментов фолатного цикла MTHFR, MTR и MTRR. 2 Дефицит витаминов: фолиевой кислоты, витаминов В6 и В12, участвующих в обмене фолатов. Генетические дефекты ферментов приводят к снижению их функциональной активности, нарушению фолатного цикла, накоплению гомоцистеина в клетках и повышению уровня гомоцистеина в плазме крови, который оказывает выраженное тромбофилическое, токсическое, атерогенное действие и обусловливает повышенный риск развития ряда патологических процессов: * Осложнения беременности (фетоплацентарная недостаточность, преждевременная отслойка нормально расположенной плаценты, поздний гестоз) * Пренатальная смерть и дефекты развития плода (незаращение нервной трубки, анэнцефалии, деформации лицевого скелета) * Сердечно-сосудистые заболевания (ишемическая болезнь сердца, атеросклероз, инфаркт миокарда, тромбозы) * Канцерогенез (колоректальная аденома, рак молочной железы и яичника) и усиление побочных эффектов при химиотерапии * Эктопия хрусталика * Остеопороз
Показания для назначения
1. Акушерская патология: привычное невынашивание, антенатальная гибель плода, поздние гестозы, преждевременная отслойка плаценты, неудачные попытки ЭКО, синдром задержки внутриутробного развития плода, рождение ребенка с изолированными пороками нервной трубки, сердца или урогенитального тракта. 2. Плановая подготовка к беременности при отягощенной наследственности (с акушерской патологией и сосудистым тромбозом в анамнезе, наличие родственников первой степени родства с наследственной тромбофилией и сердечно-сосудистыми катастрофами, особенно в раннем возрасте и др.) 3. В гинекологии: планирование или применение гормональной контрацепции, гормональной заместительной терапии у женщин, имеющих тромбозы в анамнезе и/или родственников первой степени родства с наследственной тромбофилией и тромбоэмболическими осложнениями, при планировании гинекологических операций. 4. Наличие сосудистого тромбоза в анамнезе: единичный тромбоз до 50 лет, повторные тромбозы, тромбозы необычной локализации (портальные, брыжеечные, мозговые вены), случаи наследственной тромбоэмболии и тромбоза в любом возрасте. 5. Сердечно-сосудистые заболевания: ишемическая болезнь сердца, артериальная гипертензия, атеросклероз и др. 6. Ситуации высокого риска: повышенный уровень гомоцистеина в крови (гипергомоцистеинемия), массивные хирургические вмешательства - перед трансплантацией, эндопротезированием и др., длительная иммобилизация, семейная предрасположенность к онкологическим заболеваниям и назначение химиотерапии.
Маркер
Маркер генетических мутаций фолатного цикла.
Клиническая значимость
Исследование Полиморфизм генов Фолатного цикла (MTHFR, MTR, MTRR) проводится для диагностики причины и риска развития тромбофилии, связанной с нарушением обмена фолиевой кислоты и гипергомоцистеинемией. Анализ полиморфизмов в генах фолатного цикла позволяет определить предрасположенность к опасным для жизни заболеваниям, связанным с нарушением обмена фолиевой кислоты, гипергомоцистеинемией и дает возможность своевременно назначить корректирующую терапию
Состав показателей:
Полиморфизм MTHFR 1298 A>C
Метод: Полимеразная цепная реакция в реальном времениДиапазон измерений: 300
Референтные значения:
Возраст
Комментарии
Полиморфизм MTHFR 677 C>T
Метод: Полимеразная цепная реакция в реальном времениДиапазон измерений: 300
Референтные значения:
Возраст
Комментарии
Полиморфизм MTR 2756 A>G
Метод: Полимеразная цепная реакция в реальном времениДиапазон измерений: 300
Референтные значения:
Возраст
Комментарии
Полиморфизм MTRR 66 A>G
Метод: Полимеразная цепная реакция в реальном времениДиапазон измерений: 300
Референтные значения:
Возраст
Комментарии
Выполнение возможно на биоматериалах:
Биологический материал
Условия доставки
Контейнер
Объем
Цельная кровь
Условия доставки:
24 Час. при температуре от 2 до 8 градусов Цельсия
Контейнер:
Система с ЭДТА без разделительного геля
Объем:
2 Миллилитров
Правила подготовки пациента
Стандартные условия: утром до 11.00, натощак, через 8-12 часов периода голодания. Особой подготовки не требуется. Важно: !!!Не курить в течение 30 минут до сдачи крови.
Вы можете добавить данное исследование в корзину на этой странице
Интерференция:
- Не обнаружена.
- Не обнаружена.
Интерпретация:
- Генетическое исследование выявляет в определенном участке ДНК замену нуклеотида, приводящее к замене аминокислотного состава белка и соответственно изменению биохимических свойств фермента.В результате нуклеотидных замен в кодирующем гене фермента снижается его функциональная активность. Выявляют несколько вариантов генотипа (сочетание аллелей гена), влияющих на изменение функции кодируемого им фермента: аллель «нейтральный» – нормальная активность фермента, «гетерозигота по мутантному аллелю» - сниженная активность фермента, «гомозигота по мутантному аллелю» - значительно сниженная активность фермента. Сниженная активность фермента приводит к нарушению метаболического пути превращения гомоцистеина, увеличению его содержание в плазме крови (гипергомоцистеинемии) и вероятности развития патологических состояний.