Кровь на фку у новорожденных что это такое
Скрининг новорожденных в роддоме
Ожидание рождения ребенка – период и радостный, и волнительный одновременно. Будущие родители испытывают тревогу по поводу здоровья еще не родившегося малыша. Когда чудо свершилось и маленький появился внешне здоровым, тогда все испытывают неимоверное облегчение. Но, дабы окончательно убедиться в отличном состоянии ребенка, у него берут первый в жизни анализ – скрининг.
Понятие скрининга. Выявление скрытых заболеваний
Скрининг или «пяточный тест» — это анализ крови из пятки новорожденного. Его делают в обязательном порядке во многих странах. И в его проведении имеется серьезный смысл. Он определяет наличие наследственных заболеваний.
Процедура проведения скрининга утверждена на государственном уровне, так как является наиболее эффективным методом диагностирования отклонений у детей, в том числе новорожденных. Он помогает выявлять недуги, которые были скрыты во время беременности по различным причинам. Чем раньше будет обнаружена болезнь, тем успешнее будет лечение. Ну, а если скрининг ничего не показал, то мама может даже и не знать о том, что ее малыш сдавал анализ.
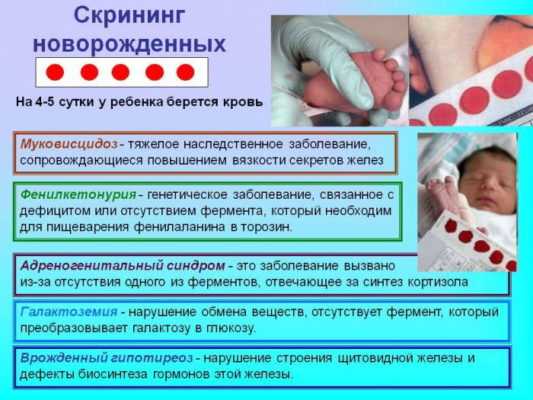
Благодаря проведению такого анализа, как пяточный тест, можно выявить следующие заболевания и отклонения у новорожденных.
- фенилкетонурия;
- муковисцидоз;
- галактоземия;
- адреногенитальный синдром;
- врожденных гипотиреоз.
Никакое другое обследование не способно выявить эти нарушения у ребенка на ранней стадии, поэтому проведение скрининга новорожденных проводится во всех родильных домах на 3-5 сутки после появления крохи на свет.
Скрининг новорожденных для обнаружения наследственных заболеваний
Анализ крови из пятки новорожденного покажет наличие или отсутствие наследственных заболеваний. Как правило, диагностируют пять наиболее распространенных патологий развития. Эти болезни связаны с нарушениями обмена веществ и при своевременной диагностике могут быть излечимы.
Фенилкетонурия (ФКУ) – это генетическое заболевание, которое возникает при нарушении расщепления финилаланина. Это одна из 20 незаменимых аминокислот, которые входят в состав белков. Она не вырабатывается самостоятельно и поступает извне с пищей. На конечном этапе финилаланин превращается в тирозин, который является медиатором. Он отвечает за скорость передачи электрических импульсов между нервными клетками.
При фенилкетонурии, поступающий финилаланин не расщепляется, накапливается в крови, происходит тяжелое токсическое отравление, страдает головной мозг и центральная нервная система. Это приводит к тяжелому умственному и физическому недоразвитию и является причиной ранней детской инвалидности. Ребенок рождается без симптомов и на первом этапе жизни болезнь трудно обнаружить.
Около 6 месяцев фенилкетонурия проявляет себе следующими симптомами:
- блуждающий взгляд;
- отсутствие реакции на голос или движение матери;
- слабая моторика;
- отсутствуют попытки перевернуться на животик или сесть.
Со временем симптомы могут становиться более выраженными, если пища, принимаемая малышом, имеет животный белок. Больной ребенок рождается в той семье, где родители носят ген фенилкетонурии.
Если обнаружить патологию на ранней стадии, исключить из питания младенца лактозу и соблюдать специальную диету, то можно прожить здоровую, долгую и счастливую жизнь.
Муковисцидоз (с латыни – вязкая слизь) – внутреннее наследственное заболевание, которое поражает легкие, потовые железы и желудочно-кишечный тракт, вызывает хронические респираторные и пищеварительные болезни. Возникает заболевание при мутации гена, который контролирует секреторные механизмы (секрет – биологически активная жидкость, выделяемая клеткой). В итоге сгущаются секреты экзокринных желез (сальные железы, потовые, молочные, слюнные, железы ЖКТ, печень), что приводит к одновременному заболеванию органов.
В первые несколько дней после рождения можно уже диагностировать муковисцидоз, наблюдая за первым в жизни актом дефекации младенца. Если ребенок самостоятельно не может избавиться от первородного кала, то нужно срочно делать анализ.
Симптомы болезни у маленьких детей:
- замедленное физическое развитие;
- сухость кожи;
- увеличенная печень;
- кожа становится землистого оттенка;
- стул – густой, с неприятным запахом;
- вздутый животик;
- затрудненное дыхание из-за слизи в легких.
Диагноз, как правило, подтверждается до двух лет. Больным необходимо постоянное наблюдение у врача и употребление медикаментов каждый день и до конца жизни. Современная медицина дает обнадеживающие прогнозы для людей с муковисцидозом.
Галактоземия – наследственное нарушение в активности фермента, который перерабатывает галактозу (простой сахар, входящий в состав молока). В печени этот моносахарид превращается в глюкозу с помощью фермента. Его отсутствие приводит к накоплению галактозы, отравлению организма и к тяжким последствиям со стороны ЦНС, глаз, ЖКТ и вызывает такие симптомы:
- сильная рвота и понос после кормления;
- отказ от пищи;
- уменьшение массы тела;
- желтуха, повышенный билирубин;
- судороги, нарушение движений.
Диагностировать заболевание можно еще в утробе мамы, но и скрининг новорожденного тоже достаточно эффективен, так как выявление патологии на ранней стадии может значительно облегчить протекание недуга и уменьшить его последствия. Патология возникает при условии, что оба родителя носят дефективный ген и вероятность передачи болезни потомству составляет лишь 25%. Молоко исключается из рациона человека навсегда. В это случае младенец не получает грудное молоко или молочные смеси, а специально разработанные адаптированные витаминные смеси.
Адреногенитальный синдром (АГС) – наследственная мутация генов, которые отвечают за выработку гормонов надпочечниками. Происходит чрезмерное продуцирование андрогенов (мужских гормонов). При этом процессе понижается количество кортизола (гормона, отвечающего за метаболизм, жировой обмен, рост, иммунитет) и альдостерона (поддерживает водно-минеральный баланс в организме, кровяное давление). Болезни подвержены мальчики и девочки в равной степени. АГС имеет три формы проявления:
- Верильная — увеличенные размеры гениталий при рождении, низкорослость, ранее половое созревание (2-4 года).
- Сольтеряющий вид приводит к обильной рвоте, диарее, гипотонии. Нарушается солевой баланс, страдает сердце.
- Гипертоническое проявление болезни встречается редко и угрожает почечной недостаточностью, кровоизлияниями в мозг, нарушением зрения.
Если оба родителя носят мутированный ген, то возможность его передачи ребенку составляет 0-25%. АГС лечится как оперативно, так и медикаментозно. Болезнь может не отражаться на жизни человека при правильном подходе.
Врожденный гипотиреоз – это ряд заболеваний, при которых щитовидная железа частично или полностью теряет свои функции, имеет нарушения в строении, не вырабатывает нужное количество гормонов для нормального развития малыша. Недуг может быть, как наследственным, так и возникать во время внутриутробного формирования. Много зависит от здоровья матери, ее образа жизни при беременности. Симптомы гипотиреоза видны сразу после рождения:
- вес ребенка свыше 4 кг;
- общая недоразвитость при сроке 40 недель и выше;
- желтоватый оттенок кожных покровов;
- мышечная слабость;
- холодные конечности;
- отечность тела;
- пупочная грыжа.
Гипотиреоз развивается постепенно. Уже к 6 месяцам он имеет ярко выраженную клиническую картину: кожа – серо-желтая, сухая, постоянные запоры, апатия, заторможенность моторики, уменьшено потоотделение, голос малыша – хриплый и низкий. В дальнейшем, у деток наблюдается интеллектуальное отставание в развитии. У девочек развитие болезни более вероятно, чем у мальчиков. Лечение, начатое в первый месяц жизни младенца, в большинстве случаев имеет огромный успех. Такие дети ничем не уступают здоровым сверстникам.
Механизмы проведения обследования
Перед отправкой мамы и ребенка домой из роддома, у малыша берут анализ на выявления наследственных и врожденных заболеваний. Забор крови делают из пятки на специальную бумагу (тест-бланк). Пять кровяных отпечатков высушивают и отправляют в лабораторию на диагностику.
При обнаружении каких-либо отклонений от нормы, о результатах скрининга сообщают родителям и приглашают на повторное обследование, чтобы окончательно удостовериться в диагнозе и при необходимости провести ряд дополнительных анализов. Скрининг – это быстрый способ выявления патологий на ранних этапах жизни ребенка.
Если повторных пяточный тест подтвердил предыдущий результат, родителей ребенка приглашают на консультацию к врачу-генетику, который в свою очередь после детальной беседы, даст направления к более узким специалистам. Ребенку будут сделать дополнительные необходимые анализы и назначено лечение.

Сроки и условия проведения скрининга
Пяточный анализ берут, как правило, на 3-5 день от рождения младенца. Ребенок должен быть доношенным и рожденным в срок. При преждевременном появлении маленького человека, скрининг делают через 7 -10 суток.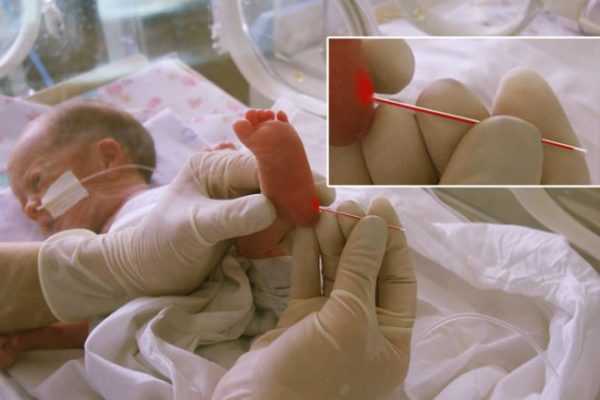
Раньше указанных сроков сдавать кровь не имеет смысла, так как результаты могут быть обманчивыми и диагноз будет неверным. Если мать с малышом были выписаны раньше четырехдневного срока, то скрининг нужно провести в поликлинике. Результаты будут доступны через 10 дней у педиатра. Ранняя диагностика болезней повышает шансы на здоровую жизнь.
что такое, как и когда делают и где можно пройти процедуру?
Главный вопрос, который волнует родителей новорожденного малыша, — здоровье ребенка. Многие врожденные болезни коварны и никак себя не проявляют на первом году жизни. Именно по этой причине был разработан метод исследования, известный как скрининг новорожденных. Это несложный тест, позволяющий выявить наличие многих врожденных заболеваний в первые дни жизни ребенка.
Что такое скрининг новорожденных
Скрининг новорожденных — это анализ крови, позволяющий провести раннюю диагностику как минимум 50 врожденных заболеваний. Метод является самым точным на сегодняшний день способом ранней диагностики генетически обусловленных патологий. По рекомендации ВОЗ скрининг новорожденных включен в перечень обязательных медицинских тестов для малышей.
На заметку
В России многие мамы называют скрининг новорожденных «пяточным тестом», поскольку кровь для него берут из пяточки малыша.
Следует знать, что положительный результат далеко не всегда означает, что у ребенка действительно есть то или иное заболевание. Для полной уверенности родителям выдадут направление на дополнительное обследование у соответствующего специалиста.
Когда проводится неонатальный скрининг у новорожденных
Это исследование проводится в первые 10 дней жизни ребенка, но точная дата может существенно меняться в зависимости от обстоятельств.
Скрининг новорожденных не рекомендуется делать в первые 2–3 дня жизни, поскольку при исследовании, проведенном так рано, есть риск ошибки — результат может быть ложноотрицательным или ложноположительным.
Обычно кровь на анализ у малышей, родившихся в срок, берут в роддоме на 4-ые сутки жизни. Недоношенным детям тест проводят на 7-ой день. Если мама с младенцем к этому времени уже выписалась из родильного отделения, кровь для скрининга берут на дому или в поликлинике.
Такие ранние сроки проведения анализа объясняются тем, что некоторые генетически обусловленные болезни проявляются в первые же недели жизни и крайне важно вовремя диагностировать их, чтобы начать терапию. Однако расширенный тест, включающий диагностику большего количества заболеваний, можно проводить и позже. Начиная с трехмесячного возраста кровь для анализа берут уже не из пятки, а из пальца.
Какие исследования входят в обязательную программу скрининга на наследственные заболевания
Поскольку болезней, которые могут быть выявлены посредством этого исследования, очень много, список патологий при обязательном скрининге сокращен до пяти самых распространенных:
- Гипотиреоз . Патология щитовидной железы, которая может привести к отставанию в физическом и психическом развитии. На сегодняшний день своевременно диагностированный гипотиреоз хорошо поддается гормональной терапии. Распространенность заболевания — 1 случай из 5 тысяч.
- Андрогенитальный синдром . Патология коры надпочечников, при которой нарушается нормальная выработка гормона кортизола. Может проявиться в виде задержки развития половой системы, проблем с сосудами и сердцем. Полному излечению этот синдром не поддается, но его можно держать под контролем при помощи гормональной терапии. Распространенность заболевания — 1 случай из 15 тысяч.
- Муковисцидоз . Заболевание проявляется заметным сгущением секрета в пищеварительном тракте и легких, что приводит к поражениям печени, ЖКТ, дыхательной системы и других органов. Поддается лечению. Распространенность заболевания — 1 случай из 3 тысяч.
- Фенилкетонурия . Заболевание, которое характеризуется нарушением выработки определенных ферментов. Последствия достаточно тяжелые. В первую очередь к ним относятся поражения ЦНС. Однако их можно избежать при помощи специальной диеты. Распространенность заболевания — 1 случай из 15 тысяч.
- Галактоземия . Так называют недостаток фермента, расщепляющего галактозу — один из сахаров, который содержится в лактозе и иных веществах. Последствия нехватки этого фермента проявляются через несколько недель жизни. У ребенка начинается желтуха, рвота, потеря аппетита. Со временем развиваются тяжелые патологии печени, замедляется умственное и физическое развитие, ухудшается зрение. Эта врожденная патология опасна, при этом встречается достаточно редко. Распространенность — 1 случай из 30 тысяч.
Данные заболевания были выбраны еще и потому, что при ранней диагностике их возможно вылечить или, по крайней мере, значительно облегчить последствия. Однако список патологий, которые можно определить при скрининге, намного шире, и при желании родители могут провести дополнительные тесты. В некоторых странах скрининг новорожденных включает диагностику большего количества заболеваний, например, в США — 40, а в Германии — 14.
Как подготовить малыша к исследованию
Чтобы результаты были максимально точными, кровь следует сдавать натощак, через 3 часа после последнего кормления. Анализ обычно проводят как минимум спустя 4 дня после начала грудного вскармливания. Подготовка к скринингу новорожденных очень проста. Перед забором крови ножку ребенка моют с мылом, протирают спиртом и насухо вытирают стерильной салфеткой — этим подготовительные меры и ограничиваются.
Как проводится забор крови
Эта процедура представляет собой довольно простой комплекс действий. На пяточке ребенка делается маленький прокол. Первая капля крови снимается стерильной салфеткой. Затем медсестра слегка сдавливает пяточку малыша и наносит полученную кровь на специальный бумажный тестовый бланк так, чтобы кровь пропитала пористую бумагу насквозь. В него вносится вся информация о новорожденном, а также об учреждении, где проводился забор крови. Бланк высушивается при комнатной температуре в течение 2–4 часов, помещается в конверт и отправляется в лабораторию или медико-генетический центр.
Интерпретация результатов анализа
На обработку образцов уходит 10–14 дней, после чего родители получают заключение генетической экспертизы. Результаты может интерпретировать только специалист. Помните, что результаты скрининга новорожденных — это еще не диагноз. Заключение может дать только врач соответствующего профиля на основании дополнительных исследований.
Несмотря на то, что скрининг новорожденных довольно точен, иногда он дает ложноположительные или ложноотрицательные результаты (чаще всего это связано с нарушением техники забора крови). Если результат анализа на какую-то болезнь положительный, родителям предлагают провести повторный тест. В случае повторного положительного результата малыш направляется на детальное обследование.
Полезная информация
Чаще всего ложноположительный результат при скрининге новорожденных дает тест на муковисцидоз.
Что такое расширенный неонатальный генетический скрининг методом ТМС?
Несмотря на то, что в программу обязательного скрининга новорожденных входит лишь пять заболеваний, генетически обусловленных болезней гораздо больше — около 500. К счастью, большинство врожденных патологий — большая редкость. Однако многие сознательные родители хотят получить как можно более полную информацию о здоровье ребенка и проходят расширенный неонатальный скрининг. Он позволяет выявить врожденные нарушения метаболизма в первые же недели жизни малыша.
Такой скрининг проводится методом тандемной масс-спектрометрии (ТМС) и дает возможность протестировать ребенка на 37 генетических заболеваний, среди которых — лейциноз, метилмалоновая ацидемия, недостаточность биотинидазы, аргининемия и множество других болезней. Из обязательного списка в такое исследование входит только фенилкетонурия.
Техника забора крови для расширенного скрининга ничем не отличается от порядка действий при плановом исследовании. Результаты анализа можно получить через 2–3 недели.
Расширенный неонатальный скрининг выявляет изменение концентрации метаболитов в ту или иную сторону, то есть повышенное или пониженное содержание этих веществ может говорить о наличии генетического заболевания. Например, при фенилкетонурии повышен фенилаланин.
Как и при обязательном скрининге, при серьезных отклонениях от нормы врач направляет малыша к узким специалистам для проведения дополнительных исследований и разработки схемы лечения, если диагноз подтвердится.
Генетический скрининг новорожденных особенно необходим, если раньше в семье были случаи наследственных заболеваний, пусть и в отдаленном прошлом. Довольно часто здоровые родители все же являются носителями дефектных генов и могут передать их потомству. Но даже если в вашей семье никто не страдал от генетических болезней, такой расширенный скрининг новорожденных сделать все равно стоит, поскольку риск наличия данных патологий у ребенка все равно есть.
Фенилкетонурия (ФКУ) у детей - симптомы болезни, профилактика и лечение Фенилкетонурии (ФКУ) у детей, причины заболевания и его диагностика на EUROLAB
Что такое Фенилкетонурия (ФКУ) у детей -
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
|
Название формы |
Уровень фенилаланина |
|
классическая |
выше 20 мг% (1200 мкмоль/л) |
|
средняя |
10,1–20 мг% (600–1200 мкмоль/л) |
|
легкая |
до 8 мг% (480 мкмоль/л) |

Что провоцирует / Причины Фенилкетонурии (ФКУ) у детей:
Основная причина фенилкетонурии у детей – нехватка фермента с названием фенилаланин-4-гидроксилаз. Из-за его отсутствия происходит скопление в жидкостях и тканях фенилаланина в большом количестве, который воздействует токсически на ЦНС ребенка. Результатом становится нарушения метаболизма гормонов, обмена белков, транспорта аминокислот, обмена глико- и липопротеидов.
Причиной ФКУ у детей может стать:
- хронический алкоголизм матери и/или отца
- инфекционные воспалительные процессы половых органов родителей
- влияние на организм отца и матери пагубных факторов и пр.
Патогенез (что происходит?) во время Фенилкетонурии (ФКУ) у детей:
Основа патогенеза фенилкетонурии (ФКУ) у детей является нехватка фермента фенилаланин-4-гидроксилаза, который обеспечивает превращение фенилаланина в тирозин. Производные фенилаланина (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты, фенилэтилламин, фенилацетилглютамин) и само вещество скапливаются в тканях. Следствием является негативное влияние на центральную нервную систему. Возникают разные нарушения, в том числе в обмене катехоламинов и серотонина.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Лечение Фенилкетонурии (ФКУ) у детей:
Новые методы лечения
На сегодня исследователи разрабатывают несколько методов альтернативной терапии фенилкетонурии (ФКУ):
- энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой
- метод «больших нейтральных аминокислот»
- лечение тетрагидробиоптерином (Сапроптерин)
Существует информация о случаях, когда пациентам с умеренной или легкой формой заболевания помогал тетрагидробиоптерин в дозе от 10 до 20 мг на 1 кг тела в сутки. В 2008 году было доказано, что для нормального физического развития детей с фенилкетонурией можно применять пищевые гликомакропептиды, которые также снижают содержание в плазме крови и головном мозге фенилаланина. Экспериментальным методом считается введение непосредственно в пораженные клетки печени ребенка введение гена фенилаланингидроксилазы. Этот метод пока не актуален в странах СНГ, в том числе Украине и России.
Продукты для детей с ФКУ (фенилкетонурией)
Диетотерапия позволяет предотвратить интеллектуальный дефицит при классической форме рассматриваемой болезни. Важное значение имеет возраст малыша, когда начинают применять диету. Каждый месяц без применения диеты малыш с ФКУ теряет примерно 4 балла IQ. Вопрос о диете при фенилкетонурии у детей в различных странах рассматривают по-разному. Но принципы едины.
При уровне уровень фенилаланина в крови до 2–6 мг% (120–360 мкмоль/л) у младенцев диета не применяется. Суть питания детей с ФКУ – в продуктах с низким содержанием фенилаланина, в основном это небелковая пища. Она нужна детям первого года жизни. В более позднем возрасте такое питание приносит меньше результатов.
Лечебный рацион питания при ФКУ:
- натуральные продукты питания
- лечебные продукты
- малобелковые продукты на основе крахмала
Детям с фенилкетонурией нельзя:
Смеси детям с ФКУ нужны только с минимальным содержанием белка. В течение первого года жизни допустимое количество фенилаланина составляет от 90 до 35 мг/кг ребенка. 50 мг фенилаланина = 1 г белка.
Лечебные продукты при фенилкетонурии у детей:
- ХР Аналог LCP
- MD мил ФКУ-0
- Афенилак
Диеты ребенку нужно придерживаться, если показатель фенилаланина в крови составляет минимум 360–480 ммоль/л.
Прикорм при фенилкетонурии (ФКУ у детей)
По достижению ребенком 3-месячного возраста рацион нужно расширять, вводя фруктовые и ягодные соки. Сначала это 3-5 капель, потом объем увеличивают до 30–50 мл. Для детей 12 месяцев доза уже составляет до 100 мл.
Соки в качестве прикорма:
- грушевый
- яблочный
- сливовый
Также в рацион постепенно вводят фруктовое пюре, постепенно увеличивая порцию. Детям от 4-4,5 месяцев уже можно овощное пюре, которое готовится родителями. Также можно плодовоовощные консервы для грудничков, но без молока. Второй прикорм – каша (10%) из безбелковой крупки или саго. Также ребенку можно давать безмолочные каши промышленного производства из кукурузной и/или рисовой муки. В них должно быть меньше 1 грамма белка на 100 мл готового продукта.
Детям от 6 месяцев можно вводить в диету кисели и/или муссы, в которых нет белка. Их готовят на амилопектиновом набухающем крахмале и фруктовом соке; это низкобелковый молочный напиток PKU «Лопрофин» и безбелковый напиток с молочным вкусом Нутриген. Детям с ФКУ от 7 месяцев можно давать низкобелковые изделия «Лопрофин»: рис, спагетти, спиральки. С 8 месяцев при фенилкетонурии малышам можно давать специальный безбелковый хлеб.
Диета для ФКУ у детей от 1 года
Для питания таких пациентов применяют продукты на основе смесей аминокислот без содержания фенилаланина и/или гидролизатов белка или с мизерным его количеством. В их составле должны быть комплексы макро-, микроэлементов и витаминов. По мере взросления ребенка дозу белка можно увеличивать, но не сразу. Количество углеводов и жиров нужно постепенно снижать, а потом и вовсе исключить. Рацион постепенно расширяется за счет натуральных продуктов и блюда.
Для детей с ФКУ от 12 месяцев можно применять специализированные лечебные продукты:
- Тетрафен 40
- Тетрафен 30
- MD мил ФКУ-1
- Тетрафен 70
- MD мил ФКУ-3
- Изифен
- П-АМ 1, П-АМ 2, П-АМ 3
- ХР Максамум (вкус нейтральный или апельсиновый)
- ХР Максамейд
Врачи советуют постепенно переходить на продукты для детей более старшего возраста на протяжении 1-2 недель. Объем предыдущей смеси нужно уменьшить на 1/4–1/5 и добавить эквивалентное по белку количество нового продукта. В части лечебных продуктов содержатся полиненасыщенные жирные кислоты. Среди малобелковых продуктов зарубежного производства есть напитки без белка, десерты, соусы и приправы, печенье и специальные сорта хлеба, которые можно детям при фенилкетонурии (ФКУ).
Часть исследователей склоняется к мнению, что детям с ФКУ диету нужно обогащать тирозином. Лечебные продукты имеют специфический вкус, потому могут быть нужны вкусовые добавки без содержания белка. Нельзя применять подсластитель аспартам, потому что при расщеплении он образует в том числе фенилаланин.
При лечении нужен регулярный контроль содержания фенилаланина в крови. Детей до 3 месяцев проверяют 1 раз в неделю, а после получения стабильных результатов – минимум 1 раз в 2 недели. Детей с ФКУ от 3 мес. до 1 года проверяют 1 раз в месяц, иногда – 2 раза. Для детей от 1 до 3 лет осмотры нужны минимум 1 раз в два месяца, а после трех лет контроль проводят 1 раз в 3 месяца.
Необходим контроль таких показателей для детей с ФКУ:
- физическое и интеллектуальное развитие ребенка
- нутритивный статус больного
- эмоциональное развитие
- речевое развитие
Один раз в месяц нужно проводить общий анализ крови. А по показаниям – биохимический анализ крови.
Если у ребенка обнаруживают дополнительные болезни с диспепсическими явлениями, интоксикацией, гипертермией, то диету можно прекратить на 2-3 дня, заменив лечебные продукты на натуральные, в которых не слишком много белка. Когда острая фаза болезни заканчивается, в рацион снова вводят лечебные продукты, но быстрее, чем в начале ввода диеты.
Прекращение диетотерапии
Возраст прекращения специальной диеты при ФКУ у детей до сих пор в процессе дискуссии. Существует информация, что при отмене диетотерапии в 5-летнем возрасте у одной трети детей с ФКУ отмечалось снижение уровня IQ на 10 баллов и более на протяжении следующих 5 лет. Прекращения диеты для детей старше 15 лет в некоторых случаях сказывались на прогрессирующих изменениях белого вещества мозга, что было выявлено при помощи МРТ.
При классической фенилкетонурии у детей диеты нужно придерживаться всю жизнь. Общее количество белка после наступления совершеннолетия не должно быть больше, чем 0,8–1,0 г/кг в сутки.
Профилактика Фенилкетонурии (ФКУ) у детей:
Чтобы организовать раннюю диетотерапию и избежать тяжелых церебральных повреждений, нужно проводить массовые скрининги на фенилкетонурию в неонатальном периоде. Это позволяет также избежать нарушения функционирования печени ребенка. Чтобы оценить риск рождения ребенка с рассматриваемым диагнозом, нужно предварительное генетическое консультирование для пар, у которых уже есть ребенок с фенилкетонурией (ФКУ) или у которых есть родственники с такой болезнью.
Женщины с фенилкетонурией до момента зачатия должны строго придерживаться диеты и продолжать ее, пока будут беременными. Это позволит избежать нарушений развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Дети с ФКУ должны наблюдаться участковым педиатром и психоневрологом.
К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
Педиатр
Психоневролог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Фенилкетонурии (ФКУ) у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни ребенка (педиатрия):
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.зачем и как его проводят

Каждую маму после рождения ребенка волнует состояние его здоровья. Различные внешние патологии можно заметить сразу, но вот большинство генетических заболеваний могут никак не проявляться в первый год жизни. Поэтому в роддомах было введено исследование — скрининг новорожденных, которое может обнаружить некоторые врожденные заболевания в первые дни жизни. Это позволит вовремя начать терапевтические мероприятия, чтобы полностью излечиться, либо хоть немного повысить качество жизни ребенка в будущем. Рассмотрим подробнее, что такое скрининг, как его проводят и какие наследственные заболевания выявляет.
Скрининг (просеивание) — анализ крови, позволяющий диагностировать у новорожденного определенное врожденное заболевание. На данный момент это исследование считается самым точным. Такая ранняя диагностика позволяет предотвратить развитие серьезных осложнений и инвалидности.
Скрининг новорожденных имеет народное название — «пяточный тест», так как забор крови происходит из пятки. Только так можно получить достаточный объем крови для исследования.
Положительный анализ не свидетельствует о том, что у ребенка имеется выявленное генетическое заболевание. Его направят к врачу для проведения более детального обследования.
Можно ли отказаться от анализа
 Мама может отказаться от проведения скрининга новорожденному. Для этого достаточно написать заявление. Неприятностей от этого никаких не возникнет.
Мама может отказаться от проведения скрининга новорожденному. Для этого достаточно написать заявление. Неприятностей от этого никаких не возникнет.
Отказавшись от генетического исследования, мама всю ответственность за здоровье своего ребенка берет на себя. Но перед принятием такого решения стоит несколько раз подумать и оценить потенциальный вред, пользу, которые такой отказ может принести.
Проведение генетического скрининга ребенку особенно нужно, если в семье диагностировались случаи врожденных заболеваний, даже у дальних родственников. Часто бывает, что здоровые родители являются носителями бракованных генов, которые могут передать детям.
Когда проводится анализ
Скрининг делают новорожденному в первую неделю после рождения, в зависимости от обстоятельств. У доношенных детей забор крови на фильтровальный бланк осуществляется в роддоме на 3 сутки жизни, недоношенных — на 7. Раньше этого времени проводить тест не стоит, так как существует риск получения ложноотрицательного или ложноположительного результата.
Если ребенок родился не в роддоме, а, например, дома, или скрининг по какой-то причине не проводился, то сдать кровь можно в поликлинике по месту жительства.
Необходимость придерживаться раннего проведения исследования у новорожденных заключается в том, что некоторые серьезные наследственные патологии проявляются в первый месяц жизни и требуют лечения.
Также младенцам может быть проведен расширенный генетический скрининг методом ТМС, включающий диагностику большинства патологий. Его делают после возраста 3 месяцев. Кровь берут из пальца.
Какие наследственные заболевания выявляет
 Обязательный скрининг новорожденных включает следующий список самых распространенных патологий:
Обязательный скрининг новорожденных включает следующий список самых распространенных патологий:
- Муковисцидоз (кистозный фиброз) — системное генетическое заболевание, вызывающее в организме образование вязкой слизи, в результате чего происходит нарушение работы органов дыхания, печени, желудочно-кишечный тракта, экзокринные желез. При раннем выявлении муковисцидоза можно значительно уменьшить риск осложнений, инвалидности и повысить продолжительность жизни. Встречается у 1 ребенка из 7000.
- Гипотиреоз — заболевание, при котором щитовидная железа вырабатывает недостаточно тиреоидных гормонов, что сильно ухудшает развитие всех систем и органов ребенка. При отсутствии терапии в первую очередь начинает страдать нервная система, что негативно влияет на умственном развитии. Также болезнь приводит к отставанию в физическом развитии. При своевременной диагностике гипотиреоз хорошо поддается лечению. Распространенность — 1 случай из 5000.
- Фенилкетонурия — нарушение аминокислотного обмена, вызванное недостаточной выработкой в печени определенных ферментов, осуществляющих превращение фенилаланина в аминокислоту тирозин. Последствия болезни тяжелые — серьезное нарушение умственного развития, поражения ЦНС. Но их можно избежать, если фенилкетонурия выявлена на ранней стадии. Лечение заключается в соблюдении специальной диеты. Встречается у 1 малыша из 15000.
- Адреногенитальный синдром — нарушение выработки гормона кортизола корой надпочечников. Патология проявляется у ребенка задержкой полового развития, нарушением роста (после 12 лет он останавливается) и солевого обмена. Вылечить синдром невозможно, но, с помощью гормональных препаратов его можно держать под контролем. Распространенность — 1 случай из 10000.
- Галактоземия — нарушение метаболизма углеводов, характеризующееся отсутствием некоторых ферментов в детском организме, что вызывает сбой в расщеплении галактозы (вещество, поступающее с молоком). Заболевание неизлечимо, но можно облегчить его течение. Раннее выявление поможет избежать тяжелых осложнений (желтуха, поражение органов, ЦНС) и даже смерти. В основу терапии входит специализированная диета, которой придерживаются около 5 лет. В тяжелых случаях ее нужно будет придерживаться всю жизнь. Также назначаются препараты для улучшения метаболизма, витамины, кальций. Распространенность — 1 случай из 10000-15000.
В список вошли именно эти врожденные патологии, потому что при ранней диагностике их можно вылечить, либо значительно снизить риск развития возможных от них тяжелых последствий. Но количество болезней, которые может определить это исследование, намного больше, и при желании родители могут провести новорожденному расширенный скрининг.
Как подготовиться к тесту
 Кровь у младенца берется только натощак — спустя 3 часа после кормления грудью. Так результат получится более точным.
Кровь у младенца берется только натощак — спустя 3 часа после кормления грудью. Так результат получится более точным.
Как проводится забор крови
Медсестра обрабатывает пятку малыша ватой, смоченной в спиртовом растворе. Затем делает прокол глубиной 1-2 мм. Легкими надавливающими движениями начинает выдавливать кровь и наносить ее на специальный бумажный бланк. При чем она должна тщательно пропитать все 5 кружков, каждый из которых отвечает за конкретное заболевание. На бланке медсестра указывает информацию о ребенке (рост, вес, дату рождения), матери (контактный телефон, адрес проживания) и роддоме (название, номер). Затем он отправляется в лабораторию.
Обработка анализа происходит в течение двух недель. Родителей уведомляют только в случае положительного результата. Но это не значит, что уже поставлен диагноз. Заключение дает только врач после дополнительного обследования. Иногда скрининг новорожденных показывает ложноположительный результат. Часто происходит это по причине нарушения техники взятия крови. Поэтому, если анализ выявил генетическое заболевание у ребенка, то педиатр выписывает направление на сдачу теста повторно. Если результат снова положительный, то его направляют на прием к генетику или эндокринологу для проведения более детального обследования.
Чаще ложноположительный результат скрининга приходит на муковисцидоз. Но обычно при повторном прохождении теста он опровергается.
Расширенный неонатальный скрининг
Обычный анализ способен выявить только 5 генетических патологий, но их гораздо больше, и, к счастью, встречаются они редко. Некоторые родители малышу делают платный расширенный скрининг методом тандемной масс-спектрометрии (ТМС), чтобы получить полную информацию о его здоровье. Исследование может выявить или исключить генетическое заболевание из 37 возможных.
Забор крови для расширенного анализа происходит в том же порядке, что и при обычном. Результат будет известен через 2-3 недели. В случае отклонения от нормы хотя бы одного показателя, врач направит малыша на дополнительное обследование.
Аудиологический скрининг
Обследование проводится в родильном доме всем новорожденным. Оно позволяет обнаружить нарушения работы слухового аппарата на ранних стадиях, чтобы своевременно начать терапевтические мероприятия. Процедура является безболезненной и недолго длится.
Аудиологический скрининг ребенку проводят на 3 день после рождения и желательно во время сна. В ушко вставляют высокочувствительный микрофон (акустический зонд), подключенный к специальному прибору. Принцип его действия — передача сигналов в ухо и регистрация реакции волосковых клеток на них. Через минут на экране устройства высветится прошел или не прошел он тест. Отрицательный результат не является постановкой диагноза, необходимо будет также пройти дополнительное обследование.
Благодаря неонатальной диагностике можно обнаружить конкретное генетическое заболевание у новорожденного на начальных стадиях и своевременно начать лечение, чтобы он смог в дальнейшем полноценно расти, развиваться и жить. Поэтому отказываться от проведения скрининга не стоит, ведь даже здоровые родители могут быть носителями дефектных генов и легко могут передать их по наследству детям.
Просмотров: 2636.причины, симптомы, диагностика и лечение
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Фенилкетонурия
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко - врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия
Редкое заболевание фенилкетонурия (ФКУ) — одна из форм наследственных дефектов обмена аминокислот. В нашей стране частота этого заболевания невелика: один больной ребенок приходится на семь тысяч здоровых новорожденных. Малыш рождается с генетическим дефектом, из-за которого аминокислота фенилаланин, поступающая в организм с пищевым белком, не может превращаться в тирозин, как это бывает в норме. В результате фенилаланин и его производные накапливаются в тканях и органах малыша, оказывая токсическое воздействие на нервную систему. Если не диагностировать ФКУ в период новорожденности и, соответственно, ничего не предпринимать, болезнь приведет к весьма серьезным последствиям: у ребенка разовьется выраженная умственная отсталость — олигофрения, вплоть до крайней степени — идиотии. При отсутствии своевременного лечения больные на всю жизнь останутся глубокими инвалидами, так как повернуть болезнь «вспять» невозможно. Однако недаром говорится, что береженого Бог бережет. Поэтому родители каждого новорожденного должны понимать, как важно вовремя обследовать ребенка, чтобы не упустить время начала лечения в случае установления диагноза ФКУ.
Причины заболевания
Больной ребенок с ФКУ может родиться только в той семье, где родители практически здоровы, но оба являются носителями патологического задатка, в данном случае — носителями гена ФКУ.
Определить носительство гена ФКУ у родительской пары возможно при генетическом обследовании на гетерозиготное носительство, проводимое в некоторых федеральных медико-генетических центрах страны. Если в семье уже есть больной ребенок с ФКУ, носительство гена ФКУ у родителей очевидно.
Однако даже если оба родителя являются носителями гена ФКУ, их дети не обязательно будут больны. Если принять за 100% всех детей, которые гипотетически могут родиться в данной семье, можно говорить о следующем риске возникновения заболевания ФКУ:
- риск рождения больных детей с ФКУ составляет 25%;
- риск рождения детей, являющихся, подобно их родителям, носителями гена ФКУ составляет 50%;
- в остальных 25% случаев родятся здоровые дети (см. рис.).
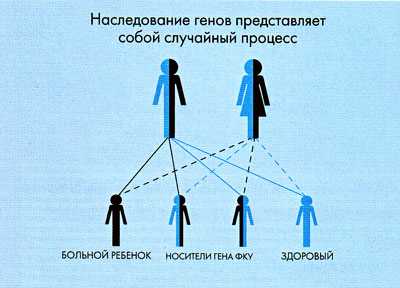
Известно, что носительство мутантного гена ФКУ среди населения составляет 2 —3%. Однако, как было сказано выше, риск рождения ребенка с ФКУ появится только в том случае, если оба родителя являются носителями патологического гена. Поэтому данное заболевание встречается довольно редко.
Симптомы
Ребенок с ФКУ рождается без каких-либо проявлений заболевания. Однако с началом кормления, при поступлении в организм белка грудного молока или его заменителей возникают первые микросимптомы, трудно распознаваемые не только родителями, но и педиатрами.
Так, в периоде новорожденности до начала лечения у ребенка с ФКУ возможны необоснованная вялость или беспокойство; обращают на себя внимание рассеянный, блуждающий взгляд, отсутствие улыбки, слабое двигательное оживление. К 6 месяцам у него выявляется задержка психомоторного развития: он перестает активно реагировать на происходящее; утрачивает способность узнавать мать; не переворачивается на живот; не пытается сесть.
Во втором полугодии жизни родители уже не могут не заметить непонимание речи взрослого, неумение выражать голосом и мимикой ребенка свои переживания. У детей старше трех лет нарастают умственная отсталость, возбудимость, повышенная утомляемость; нарушается поведение, что проявляется в расторможенности, психотических расстройствах.
Часто у нелеченых больных ФКУ моча имеет своеобразный «мышиный» запах. Иногда возникают судорожные приступы различной степени выраженности; экзематозные изменения на коже.
Диагностика
С первых дней после появления на свет здоровый ребенок должен очень быстро набирать темп развития, приобретая разнообразные физиологические навыки. Следите за его психомоторным развитием, постоянно консультируйтесь у врача-педиатра.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия - самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии.
К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних 10 лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Минздрава РФ №316 от 30.12.1993 г. организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории России. На службе у программы скрининга находятся все родовспомогательные учреждения страны, детские районные поликлиники, более 80 медико-генетических центров, консультаций и кабинетов.
Для раннего выявления ФКУ всем новорожденным проводится скрининг-тест (анализ крови ). Это самый надежный способ позаботиться о здоровье малыша с первых дней его рождения.
Скрининг-тест заключается в следующем:
- У новорожденного не позднее чем на четвертый-пятый день его жизни натощак (через 3 часа после кормления) берется несколько капель крови из пяточки.
- Кровь наносится на специальный бумажный бланк, выданный лабораторией скрининга в медико-генетическом центре региона по месту рождения ребенка. Необходимо, чтобы капли крови были нанесены на бланк тремя насквозь пропитанными кровью кружками диаметром не менее 12 мм.
- Бланк с кровью как можно быстрее должен быть возвращен в лабораторию скрининга для проведения анализа крови на содержание в ней аминокислоты — фенилаланина (ФА).
- Анализ крови на ФА выполняется через сутки после поступления бланка в лабораторию.
- Результат скрининг-теста заносится в обменную карту ребенка в роддоме в виде штампа: «На ФКУ и ВГ1 обследован».
При выписке из роддома обязательно проверьте, был ли проведен скрининг-тест на ФКУ.
Если роды происходили вне родильного дома (в обычной больнице, дома и т.д.) и скрининг-тест не производился, родителям самостоятельно без промедления следует обратиться в региональную медико-генетическую консультацию.
В Москве скрининг-тест и консультация врача-генетика проводятся в Московском центре неонатального скрининга по адресу: 119334, 5-й Донской проезд, д.21 а. Телефоны: 952-22-28, 954-41-27.
Если скрининг-тест окажется положительным, вас оповестят и пригласят на консультацию к врачу-генетику. При повторном положительном результате анализа на ФА следует немедленно начать лечение ребенка.
Диагноз ФКУ должен быть поставлен (или отвергнут) не позднее трехнедельного возраста ребенка!
Родителям не следует поддаваться панике: вероятность того, что заболевание будет обнаружено именно у вашего ребенка, очень невелика, но даже если это случится, - не волнуйтесь: развитие болезни можно предотвратить при своевременно начатом лечении.
Лечение
Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза. Смысл диетического лечения сводится к резкому ограничению поступающего с пищей белка животного происхождения и замене его специализированными лечебными продуктами. Лечебный продукт — это сухая смесь аминокислот без фенилаланина, являющаяся практически единственным источником пищевого белка в рационе, необходимым для роста и развития ребенка. Родители детей, больных ФКУ, получают лечебные продукты в медико-генетической консультации бесплатно.
В случае выявления заболевания фенилкетонурии у новорожденного родители немедленно получают квалифицированную консультативную помощь и специализированную литературу у врача-генетика в медико-генетическом центре, консультации или кабинете по месту жительства. Госпитализации ребенка не требуется.
Течение беременности у женщины, являющейся носительницей гена ФКУ ничем не отличается от течения беременности здоровой женщины.
Желаю вам благополучных родов и исполнения заветной мечты — рождения здорового ребенка.
1Аббревиатурой ВГ обозначается другое врожденное заболевание - врожденный гипотиреоз.
У ребенка 4 месяца взяли повторный анализ на ФКУ. Кто знает что это такое?
ФЕНИЛКЕТОНУРИЯ, врожденное, передающееся по наследству нарушение обмена веществ. Его причиной служит недостаточность определенного фермента, а именно фенилаланингидроксилазы, необходимой для нормального метаболизма аминокислот, из которых состоят белки. В отсутствие этого фермента не происходит превращения аминокислоты фенилаланина в другую аминокислоту – тирозин. В результате резко возрастают уровни фенилаланина в крови и фенилкетона – производного фенилаланина – в моче. Симптомы фенилкетонурии проявляются в раннем детстве и включают рвоту, шелушащуюся кожную сыпь, раздражительность и затхлый («мышиный» ) запах тела, обусловленный аномальным составом мочи и пота. Симптомы со стороны центральной нервной системы могут быть разными, обычно это навязчивые движения, поддергивания, судороги. Самое тяжелое осложнение заболевания – задержка психического развития, которая в отсутствие лечения практически неизбежна. Фенилкетонурия наследуется как рецессивный признак, что означает обязательное присутствие дефектного гена – причины данного заболевания – как у отца, так и у матери ребенка. Болезнь развивается лишь в том случае, если ребенок унаследовал оба дефектных гена. Вероятность рождения больного ребенка в семье, где оба родителя – носители дефектного гена, при каждой беременности составляет примерно 1:4. Среди белого населения США фенилкетонурия встречается с частотой ок. 1:20 000. Большинство больных – голубоглазые блондины; их кожа, глаза, волосы обычно светлее, чем у здоровых родственников. Среди темнокожего населения болезнь наблюдается редко. Анализы крови на фенилкетонурию должны проводиться у всех новорожденных еще в родильных домах. Если анализ обнаруживает высокий уровень фенилаланина, необходимы дальнейшие исследования для подтверждения диагноза. Важность предварительных анализов у новорожденных связана с тем, что, судя по имеющимся данным, проявления заболевания, особенно задержку психического развития, можно предотвратить диетой с малым количеством фенилаланина. Разработаны питательные смеси с низким содержанием фенилаланина, предназначенные для детей с этим заболеванием. P.S. В роддомах берут анализ не только не ФКУ, а так называемый неонатальный скрининг - на 5 заболеваний: ФКУ, врождённый гипотиреоз, галактоземию, адреногенитальный синдром, муковисцидоз. Но, по привычке, это называют анализ на ФКУ. Если этот анализ дал положительный (сомнительный) ответ, его повторяют для уточнения - какая именно патология из перечисленных выявлена. Подробнее про этот анализ: <a rel="nofollow" href="http://www.rost.ru/projects/health/p04/p36/a36.shtml" target="_blank">www.rost.ru/projects/health/p04/p36/a36.shtml</a> –
Фенилкетонурия - это редкое генетическое заболевание, одна из форм наследственных дефектов обмена аминокислот. Не буду рассказывать Вам всё то, что пишут в медицинских справочниках - Вы сами можете найти это всё в интернете. Лучше посмотрите этот форум: <a rel="nofollow" href="http://www.rodim.ru/conference/index.php?showtopic=22353" target="_blank">http://www.rodim.ru/conference/index.php?showtopic=22353</a> Там общаются родители, у чьих детишек ФКУ. Думаю, Вы найдёте там что-то полезное для себя. Вообще, паниковать пока подождите. Если повторный анализ (т. н. ретест) вновь демонстрирует высокий уровень ФА, подозрение на фенилкетонурию становится довольно серьезным - рекомендуется детальное обследование ребёнка, которое обычно и подтверждает либо исключает ФКУ у малыша. Вы уточните уровень фенилаланина, отмечавшийся у ребенка на скрининге и на ретестах. Именно эти цифры имеют ключевое прогностическое значение (до 2 мг% - норма, 2-8 мг% - транзиторная гиперфенилаланинемия, 8-15 мг% - гиперфенилаланинемия, требующая дополнительной диагностики, 15 мг% и выше - фенилкетонурия различной степени тяжести) . Если коротко, дообследуйте ребёнка!
Фенилкетонурия (ФКУ) - наследственное заболевание обмена веществ, без лечения (или при несвоевременном, неадекватном лечении имеющее весьма тяжелые последствия. Причиной болезни являются унаследованные от обоих родителей мутации ("поломки") гена РАН, кодирующего специфический белок-фермент - фенилаланингидроксилазу. Родители при этом, как правило, здоровы - поскольку являются гетерозиготными носителями мутантных генов РАН - т. е. один из двух генов у отца и матери нормален, а один дефектен. Дефектность этого фермента превращает довольно распространенную в пищевых продуктах аминокислоту фенилаланин (ФА) в потенциально токсичное для организма ребенка вещество - при фенилкетонурии вполне обычная пища может служить причиной грубого химического повреждения головного мозга малыша. Без соответствующего лечения, начатого в первые 3 месяца жизни, фенилкетонурия довольно быстро приводит к необратимому поражению мозга ребенка - что проявляется интеллектуальным дефицитом (умственной отсталостью) . Если же лечение фенилкетонурии начинается рано и проводится адекватно, эффект бывает весьма неплохим - ребенок зачастую избегает инвалидизации и впоследствии учится в обычной (общеобразовательной) школе.
Самое важное в этом анализе, что он должен браться ИСКЛЮЧИТЕЛЬНО на тощак! У нас его брали в роддоме ВО ВРЕМЯ КОРМЛЕНИЯ (если б я тогда знала, конечно, что-нибудь предприняла) Через какое-то время домой пришло письмо (не помню откуда, из какого-то центра, где эти анализы обработываются), в котором говорилось о том, что нам нужно пересдать этот анализ, якобы он какой-то не понятный, не хороший и т. д. И еще написано-что если вы ответа-письма больше не получите, значит все хорошо! Нам уже 2,4 и тьфу-тьфу-тьфу!
У меня оно тоже.
3 метода диагностики, основные симптомы и 4 важных правила диетотерапии
О болезни
Фенилкетонурия (ФКУ) – генетическое заболевание, которое связано с нарушением обмена аминокислот, главным образом фенилаланина. Поступление в организм малыша обычной белковой пищи приводит к накоплению в тканях токсических продуктов обмена веществ. Эти соединения необратимо влияют на нервную систему и головной мозг ребёнка, вызывают прогрессирующее слабоумие.
В некоторых странах недуг носит имя первооткрывателя и называется болезнью Фёллинга. Иное название, фенилпировиноградная олигофрения, указывает на патогенез заболевания.
Статистика распространения наследственной болезни отличается в различных странах. Так, в России недуг обнаруживается у одного малыша из 5 – 10 тысяч новорождённых (в зависимости от региона страны). Наиболее безопасной страной в отношении риска развития фенилкетонурии считается Финляндия (1:100000). Турция же занимает первое место по возникновению генетического недуга, один из 2600 новорождённых имеет нарушенный обмен фенилаланина.
Среди больных фенилкетонурией девочки встречаются в 2 раза чаще, чем мальчики. Наследование происходит по аутосомно-рециссивному типу, что означает наличие мутантного гена в обеих семьях родителей. Здоровые носители генетического дефекта могут не догадываться о своей особенности и не иметь никаких внешних проявлений. Ситуация усложняется, когда обладатели мутации вступают в брак и собираются завести детей. Риск появления на свет малыша, больного фенилкетонурией, в таком случае составляет 25%. Половина детей, рождённых в данной семье, останутся бессимптомными носителями наследственного дефекта.

Хотя заболевание может привести к грубым, необратимым нарушениям в развитии малыша и значительно ухудшить качество жизни крохи, неприятных последствий можно избежать. Фенилкетонурия – одна из немногих наследственных болезней, которая эффективно поддаётся лечению. При своевременно принятых мерах малыш практически всегда вырастает здоровым.
Историческая справка
Впервые наследственную природу слабоумия заподозрил норвежский биохимик и психиатр Ивар Асбьёрн Фёллинг. Наблюдая за группой умственно отсталых детей, врач заметил общность клинической картины и обнаружил изменение химического состава мочи больных брата и сестры. Добавив к биологической жидкости раствор хлорного железа, доктор выявил изменение окраски мочи, появление оливково-зелёного оттенка. Наличие изменений анализов у близких родственников дало основание заподозрить наследственные причины заболевания.
Этот метод обнаружения фенилкетонурии не потерял своей актуальности и в современном мире. Определение в моче фенилпировиноградной кислоты специалисты называют пробой Фёллинга в честь норвежского врача.

Как развивается болезнь?
Предугадать развитие фенилкетонурии у новорождённого малыша без применения дополнительных исследований невозможно. Кроха ничем не отличается от своих сверстников, выглядит абсолютно здоровым ребёнком. Изменения происходят внутри организма, когда начинается энтеральное вскармливание, в организм поступают белки грудного молока.
Причина патологии кроется в недостатке определённых ферментов, участвующих в превращении незаменимой аминокислоты фенилаланина в тирозин. Таким образом происходит повышение уровня фенилаланина и его производных в крови и спинномозговой жидкости. Продукты обмена веществ оказывают токсическое действие на клетки головного мозга, нарушают жировой обмен и передачу нервных импульсов между нейронами.
В то же время необходимый для нормального функционирования тирозин, не синтезируется в должном количестве. Эта аминокислота является незаменимой и принимает активное участие в образовании гормонов, нейромедиаторов, пигмента меланина.
Различают несколько клинических форм фенилкетонурии, которые отличаются дефицитом определённого фермента в печени. В 98% возникает ФКУ I типа, связанная с дефектом гена в 12-й хромосоме. Недуг проявляется недостатком фенилаланин-4-гидроксилазы и хорошо поддаётся лечению. II и III типы заболевания относятся к крайне редким недугам и характеризуются тяжёлыми проявлениями, неэффективностью диетотерапии в коррекции обмена аминокислот.
Симптомы классической формы фенилкетонурии
Общие проявления
Если неонатальный скрининг не был проведён в родильном доме, заболевание осталось не распознанным и не были организованы необходимые лечебные мероприятия, то ФКУ проявит себя на первом году жизни крохи. Первые признаки отклонений от нормы родители обнаружат у ребёнка в возрасте 2 – 6 месяцев, когда ранее здоровый карапуз становится вялым и апатичным или наоборот, чрезмерно раздражительным. Приём пищи часто заканчивается срыгиванием, а на коже младенца появляются проявления аллергического дерматита.
Характерная внешность
Поскольку нарушение обмена аминокислот приводит к уменьшению образования меланина, в случае развития заболевания кожные покровы, волосы, радужная оболочка глаз теряют пигмент. Малыши с генетическим заболеванием имеют светлые волосы, тонкую бледную кожу, голубые глаза. Указывать на наследственный недуг может характерный запах пота и мочи крохи. Многие врачи характеризуют его как «мышиный», а связан он с выделением продуктов обмена аминокислот с биологическими жидкостями.

Поражение нервной системы
Первые признаки изменений со стороны нервной системы проявляются вялостью, снижением тонуса мышц крохи. Нередко возникает дистония, появляются непроизвольные сокращения мышц, навязчивые движения, судороги. Возможно развитие органических патологий головного мозга – микроцефалии и гидроцефалии.
Практически у 50% больных фенилкетонурией развивается эпилепсия. Иногда характерные приступы являются первым признаком наследственного заболевания. Пароксизмы плохо поддаются лечению обычными противосудорожными препаратами и значительно снижают качество жизни ребёнка.
Отставание в развитии
С течением болезни малыш быстро теряет интерес к окружающему миру, становится апатичным. Физическое развитие ребёнка так же страдает – кроха поздно овладевает необходимыми навыками. Задержка в психическом развитии становится очевидной, когда возникают проблемы в формировании речи, отставание в умственном развитии.

Исследования показали, что грубые изменения интеллектуальной деятельности развиваются уже через год прогрессирования болезни. Кроха необратимо теряет до 50 баллов IQ ежегодно, что впоследствии приводит к развитию дебильности, имбецилии или идиотии. Поэтому сроки начала лечения фенилкетонурии очень важны.
Неврологические проблемы у детей старшего возраста выражаются в гиперактивности, появлении стереотипий, навязчивых движений кистей рук, лица, языка, покачиваний. Иногда расстройства психики проявляются в шизофреноподобных состояниях.
Грубое отставание в развитии приводит к отсутствию навыка ходьбы у 30% больных детей и алалии, недоразвитии речи, у 60% пациентов.
Разновидности патологии
Транзиторная форма гиперфенилаланинемии
Не всегда недуг протекает классически, иногда повышение уровня фенилаланина носит временный характер, а само заболевание не развивается. Непродолжительное увеличение концентрации аминокислоты в крови ребёнка может быть связано с незрелостью ферментных систем малыша, что наиболее характерно для глубоко недоношенных детей. Такие малыши требуют более детального обследования для определения причины заболевания.
Птерин-зависимые варианты фенилкетонурии
Примерно у 2 % малышей, у которых была выявлена фенилкетонурия в родильном доме и назначена соответствующая диета, возникают неврологические проблемы. Эти симптомы могут указывать на развитие у крохи редкой, птерин-зависимой формы ФКУ (тип II, III по классификации). Клинические проявления атипичных форм ФКУ сходно с течением классической формы, но симптомы прогрессируют быстрее, а диетотерапия не оказывает необходимого эффекта.

Огромную роль в развитии птерин-зависимых форм играет дефицит нейромедиаторов, который возникает из-за нарушения обмена аминокислот. Эти вещества необходимы для передачи нервных импульсов между нервными клетками, без которого нормальное функционирование организма невозможно.
Фенилкетонурия II считается более злокачественной, чем III типа. Грубые нарушения обмена аминокислот приводят к массовой гибели нервных клеток. Нередко малыши с прогрессирующей ФКУ II типа гибнут в возрасте 2 – 3 лет.
Материнская фенилкетонурия
Не всегда больные ФКУ дети рождаются от здоровых родителей. Если беременные женщины с наследственной патологией не соблюдают диеты, высока вероятность рождения малыша с синдромом материнской фенилкетонурии. Токсические вещества проникают через плаценту из материнского организма к плоду и нарушают формирование органов и систем ребёнка. На свет появляются дети с множественными врождёнными пороками развития, микроцефалией, умственной и физической отсталостью.
Диагностика
Медико-генетическое консультирование семьи
Будущим родителям нужно быть особенно внимательными, если в семьях кого-либо из них имели место случаи рождения больных фенилкетонурией детей. Нельзя исключить возможность появления на свет ребёнка с ФКУ, даже если недуг проявился у дальнего родственника. Коварство заболевания кроется в бессимптомном носительстве повреждённого гена абсолютно здоровыми людьми.
Всем парам, у которых имеются подозрения на наличие наследственных болезней в роду, стоит обратиться к врачу-генетику ещё во время планирования малыша. В медико-генетическом центре (МГЦ) с помощью современных методов диагностики можно выявить носительство мутантного гена и рассчитать риск рождения ребёнка с ФКУ и другими генетическими болезнями.
Инвазивные методы диагностики (хорионбиопсии, амниоцентеза, кордоцентеза) могут помочь определить недуг до рождения крохи. При этом исследуется генетический материал, полученный от плода. Данные методы являются травматичными и могут повлечь за собой спонтанное прерывание беременности. Поэтому применение их оправдано лишь при доказанном носительстве мутантных генов у родителей и высоком риске возникновения ФКУ у малыша.
Неонатальный скрининг
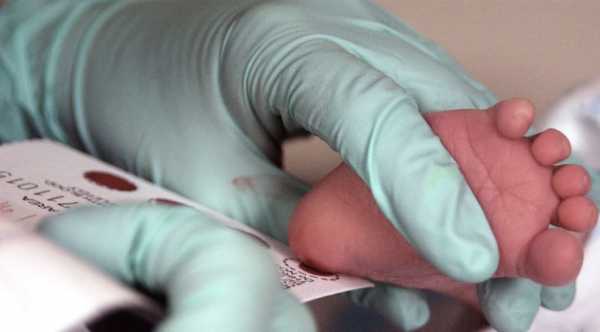
Перед выпиской из родильного дома новорождённых малышей массово обследуют на наследственные заболевания. Для этого важного исследования медработник набирает кровь из пяточки малыша и наносит капельки биологической жидкости на фильтровальную часть тест-бланка. Каждая капля крови предназначена для выявления одного из 5 заболеваний: фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии.
Неонатальный скрининг – очень важное и необходимое всем детям без исключения обследование. Он проводится совершенно бесплатно и направлен на сохранение здоровья нации. Отказываясь от манипуляции, родители совершают огромную ошибку, ведь симптомы наследственных недугов не всегда заметны у новорождённого малыша. Яркие клинические проявления нередко возникают, когда помочь ребёнку уже сложно, а изменения в организме крохи приняли необратимый характер.
Этот метод является достоверным в выявлении фенилкетонурии, если малыш получает достаточное количество энтерального питания, грудного молока. Поэтому новорождённым, которые находятся в отделении реанимации, анализ проводят позже. Отличаются и сроки постановки пробы у доношенных и недоношенных детей. Малышам, которые появились в срок, рекомендован набор капиллярной крови на 4-е сутки жизни. Проведение исследование родившимся преждевременно крохам откладывается до 7-х суток.
Анализ следует проводить натощак, что обеспечит более точный результат и повысит диагностическую значимость теста. Бланк с капельками крови малыша и паспортными данными родителей отправляется в лабораторию медико-генетической консультации, где проводится биохимическое исследование.
Родителям нужно обратить внимание на правильность заполнения бланка, корректность своих контактных данных. На проведение исследования уходит в среднем 10 дней, семья к этому времени обычно находится дома. В случае обнаружения положительного результата теста, родителям нужно будет в кратчайшие сроки обратиться в медико-генетический центр, а неправильные контактные данные задержат дальнейшее обследование малыша.
Обследование ребёнка в МГЦ
Чтобы подтвердить заболевание у младенца и установить его причину, малышу придётся пройти многоэтапную процедуру обследования. Повторные биохимические анализы помогут подтвердить повышение уровня аминокислоты в крови и принять решение о необходимости диетотерапии. Проводится исследование уровней фенилаланина и тирозина, активность печёночных ферментов, определение продуктов обмена аминокислот в моче.
Затем осуществляется молекулярно-генетическая диагностика, которая позволяет выявить мутацию в гене РАН, ответственном за развитие фенилкетонурии. С помощью данных методов можно определить и бессимптомное носительство заболевания.
Лечение ФКУ
Самым эффективным методом терапии классической формы фенилкетонурии считается правильно организованная диетотерапия. Только исключив из рациона крохи продукты, содержащие большое количество аминокислоты, можно ожидать положительных результатов от лечения.
Особенности диетотерапии
Правила подбора пищевых продуктов
Фенилкетонурия у детей – непростое заболевание и правильно подобрать рацион для малыша может только врач. Выбирая разрешённые продукты и рассчитывая их объём, доктор учитывает некоторые индивидуальные особенности:
- какая из форм болезни присутствует у малыша;
- степень тяжести недуга, уровень фенилаланина в крови;
- возраст крохи;
- особенности физического развития непоседы;
- физиологическую потребность растущего организма в белке;
- как переносит малыш поступление небольшого количества фенилаланина с пищей.
Аминокислота фенилаланин является незаменимой, а значит может поступать в организм только с белковой пищей. Растущий ребёнок нуждается в достаточном поступлении нутриентов, ведь около 40% пищевого фениаланина расходуется на выстраивание собственных белков. Получается, что полностью исключить из рациона крохи опасные продукты также невозможно. Поэтому в первые дни диетотерапии лабораторно оценивается, как организм крохи реагирует на небольшое поступление аминокислоты с продуктами питания.
Возможно ли грудное вскармливание при фенилкетонурии?
Поскольку, в большинстве случаев, болезнь определяется ещё в первые недели жизни крохи, становится вопрос о безопасности кормления малыша грудным молоком. На самом деле, грудное вскармливание необходимо для нормально развития ребёнка, становления его иммунной системы. Но применение его оправдано лишь в случаях, когда мать придерживается чётких правил и строго контролирует употребляемый малышом объём съеденной пищи.
Для безопасного кормления лучше использовать свежесцеженное грудное молоко, допустимое количество которого рассчитывает врач исходя из массы тела малыша и его потребности в фенилаланине.
Чем младше ребёнок, тем выше его потребность в аминокислоте. Например, допустимый объём фенилаланина для новорождённого малыша составляет 90 – 60 мг/кг массы тела в сутки, в то время как необходимое количество аминокислоты для школьника снижается до 10 – 20 мг/кг массы тела. Количество фенилаланина в продуктах можно рассчитать, учитывая, что 1 г белка содержит около 50 мг аминокислоты.
Специализированные смеси
Существуют специально разработанные продукты для рационального питания больных фенилкетонурий малышей. Они изготавливаются на основе гидролизатов белка или смеси аминокислот, которые полностью или частично лишены фенилаланина. Питание лечебными смесями направлено на восполнение организма крохи необходимым белком, достаточное поступление питательных веществ. Эти смеси могут использоваться как заменители грудного молока или применяться, как добавка к основному рациону для старших детей.
Состав лечебных смесей для детей разного возраста отличается. Например, для ребят до года подойдут такие продукты, как «Афенилак», «Лофеналак». Старшим детям показаны «Тетрафен», «Фенил-фри», «Максамум – ХР».

«Пищевой светофор»
С каждым годом малыш растёт, и его потребности в продуктах питания увеличиваются. Приходит время вводить прикорм, пробовать новые блюда. Родителям нужно помнить об опасности фенилаланина для мозга малыша, выбирать только разрешённые врачом продукты.
Рацион больного ребёнка значительно отличается от питания сверстников. Прикорм начинается с введения овощей и фруктов, затем следует добавлять безбелковые макароны, некоторые виды круп. Составлять первое меню малыша и контролировать содержание аминокислот в крови на фоне изменения рациона должен лечащий врач.
Все продукты разделяют на группы по содержанию в них фенилаланина:
- красный список.
Рекомендуется полностью исключить продукты этого перечня, поскольку они богаты белками. Это мясо и субпродукты, рыба, яйца, хлеб, гречневая, ячневая крупы, хлопья « Геркулес»;
- жёлтый список.
Допустимо использовать небольшое количество продуктов из этой группы под контролем анализа крови. Возможно введение кукурузной и рисовой крупы, картофеля, молочных продуктов с низким содержанием белка;
- зелёный список.
Эти продукты составляют основу питания больных фенилкетонурией детей старшего возраста: мука кукурузная и рисовая, большинство овощей, фрукты и ягоды, сладости, сливочное масло.
При применении готовых продуктов нужно внимательно читать инструкцию на упаковке. Употребление бананов, сухофруктов, бобовых может повысить концентрацию фенилаланина, поэтому их приём стоит ограничить.

Другие методы терапии
Применение медикаментов оправдано лишь для лечения птерин-зависимых форм ФКУ, когда диета не оказывает надлежащего эффекта. В таких ситуациях используют препараты L-дофы и 5-гидроокситриптофана.
В остальных случаях возможно использование витаминотерапии, минеральных комплексов, как дополнение к диете. При необходимости назначаются ноотропные препараты, антиконвульсанты. Работа с логопедами, психологами, массаж, физиотерапия так же оказывают положительный результат в лечении недуга.
Прогноз и профилактика
Течение классической формы заболевания значительно не влияет на продолжительность жизни человека. Но без надлежащего лечения тяжёлые симптомы недуга значительно сказываются на интеллекте и качестве жизни пациента. В случае рациональной диетотерапии и своевременного контроля уровня фенилаланина в крови, дети с ФКУ не отличаются от своих сверстников и хорошо адаптируются в обществе. Атипичные формы недуга нередко приводят к инвалидизации и смерти ребёнка, поскольку лечение редко оказывает хороший результат.
Специалисты рекомендуют соблюдать диету в течение всей жизни человека, но некоторые послабления возможны по достижению ребёнком 18 лет. Особенно внимательными нужно быть беременным женщинам, больным ФКУ, чтобы избежать синдрома материнской фенилкетонурии у ребёнка.
Профилактика заключается в предупреждении близкородственных браков, медико-генетическом консультировании семей из группы риска по развитию ФКУ, проведении массового обследования новорождённых.

Выводы
Болезнь у малыша – огромный стресс и испытание для молодых родителей, особенно если недуг носит наследственный характер. Но фенилкетонурия – одна из немногих генетических болезней, мероприятия по лечению которой приносят хорошие результаты.
Массовое обследование новорождённых — Википедия
Массовое обследование новорождённых (неонатальный скрининг) — один из эффективных способов выявления наиболее распространенных врождённых и наследственных заболеваний у новорождённых детей.
Позволяет обеспечить раннее выявление заболеваний и их своевременное лечение, остановить развитие тяжёлых проявлений заболеваний (фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии), ведущих к инвалидизации.
В Швеции PKU тест проводится с 60-х годов, список диагностируемых заболеваний постепенно расширялся. Диагностика бесплатна. Предлагается также для усыновленных/удочеренных детей и детей-иностранцев до 8 лет). Диагностируемые в Швеции заболевания (25 шт.):
Источник: https://www.karolinska.se/for-vardgivare/karolinska-universitetslaboratoriet/centrum-for-medfodda-metabola-sjukdomar/nyfoddhetsscreening/
И это-никакой не нац.проект, а обычная пректика. Легче/дешевле выявить на ранних сроках, чем в позднем периоде. Абсолютно бесплатно, по крайней мере, точно-для граждан. Про беженцев-не уверена.
В соответствии с международными рекомендациями в России на протяжении 15 лет[прояснить] проводится неонатальный скрининг на фенилкетонурию и врождённый гипотиреоз.
В рамках реализации приоритетного национального проекта «Здоровье»[1] с 2006 года в неонатальный скрининг начато внедрение диагностики таких заболеваний, как адреногенитальный синдром, галактоземия и муковисцидоз.
С 2007 года в перечень выявляемых заболеваний включён аудиологический скрининг детей первого года жизни, который позволит своевременно провести диагностику нарушений слуха у ребёнка и последующую реабилитацию тугоухости.
С 2012 года в Свердловской области неонатальный скрининг расширен до 16 заболеваний [2]. Исследование проводится в сухом пятне крови методом тандемной масс-спектрометрии (MS-MS). Список дополнительных 11 заболеваний:
- лейциноз
- тирозинемия типа I
- цитруллинемия
- множественная карбоксилазная недостаточность
- недостаточность очень длинных цепей ацил-СоА-дегидрогеназы жирных кислот
- недостаточность средний цепей ацил-СоА-дегидрогеназы жирных кислот
- недостаточность митохондриального трифункционального белка (недостаточность длинных цепей гидроксил-СоА-дегидрогеназы жирных кислот)
- глютаровая ацидурия типа I
- изовалериановая ацидемия
- метилмалоновая ацидемия
- пропионовая ацидемия
С 2018 года в городе Москве неонатальный скрининг расширен до 11 заболеваний[3]. В него были включены ещё 6 заболеваний:
При выборе заболеваний для неонатального скрининга, в соответствии с рекомендациями Всемирной организации здравоохранения, учитывались такие факторы, как тяжесть проявления заболеваний, частота распространения данных заболеваний, а также простота и достоверность применяемых методов диагностики, наличие доступных и эффективных средств лечения.
В США массовый неонатальный скрининг проводится с 1963 года[4].
Сроки и условия проведения обследования[править | править код]
Неонатальный скрининг начинается в родильном доме: у каждого новорождённого берется несколько капель крови на специальный тест-бланк, который направляется в специализированную лабораторию для проведения исследования.
"Проба ПКУ берется у всех новорожденных в Швеции как можно скорее после 48-часового возраста, чтобы выявить ряд редких заболеваний, при которых важно быстро начать лечение. Образец представляет собой анализ крови, который капают на специальную фильтровальную бумагу и отправляют в лабораторию для анализа."
Источник: www.karolinska.se/pku
Возможно, данная информация недостоверна: "У доношенных детей кровь для исследования берут на 4 день жизни, у недоношенных — на 7 день жизни." Источник: тот, кто редактировал до меня.
Для исследования берут периферическую кровь — из пяточки. Взятие крови производят утром, ребёнок должен быть натощак (3 часа после очередного кормления).
В случае обнаружения в крови маркера заболевания родители с новорождённым ребёнком приглашаются в медико-генетическую консультацию для проведения дополнительного обследования. Это может быть повторное исследование в сухом пятне крови, исследование в сыворотке крови, потовый тест, копрограмма, ДНК-диагностика. При подтверждении диагноза назначается лечение. В дальнейшем ведется динамическое наблюдение за ребёнком.
Фенилкетонурия - О ФКУ
У вас родился ребенок с ФКУ
Чего ожидать
В США все новорожденные проходят скрининговый тест на фенилкетонурию (ФКУ) в течение примерно 24 часов после рождения. Кровь берется из пятки новорожденных и в лаборатории определяется уровень фенилаланина (ФА) в крови. Если уровень ФА высокий, выполняются дополнительные исследования для подтверждения ФКУ. Педиатр помогает каждой семье связаться со специалистом по нарушению обмена веществ для проведения окончательных исследований. Как только диагноз определен, начинается лечение, которое заключается в снижении уровня ФА в крови до безопасного уровня, т.к. высокий уровень ФА в течение продолжительного времени может привести к повреждению мозга. С помощью скрининга новорожденных, раннего лечения и регулярного контроля за уровнем ФА, возможно сохранить уровень ФА в пределах безопасных значений.
В первые недели и месяцы возникает необходимость начать делиться опытом с другими и позволить близким членам семьи оказывать помощь. Одна из проблем, с которой сталкиваются родители ребенка с ФКУ, это то, что не многие слышали об этом заболевании. Со временем становится легче находить возможности объяснять что такое ФКУ вашей семье, друзьям, ребенку и другим заинтересованным лицам. Некоторые люди сразу после рождения ребенка готовы рассказать об этом семье и друзьям; другие предпочитают подождать, прежде чем сообщить диагноз. Вы сами должны выбрать подходящее время, когда вы будете готовы поделиться с другими этой информацией.
Вот некоторые ключевые моменты, которые вы, возможно, захотите сообщить:
- ФКУ это генетическое заболевание, которое не передается другим людям
- Кроме необходимости соблюдения специальной диеты, ваш ребенок здоров
- Организм вашего ребенка не может расщеплять аминокислоту фенилаланин (ФА), которая содержится в белковой пище
- ФА может концентрироваться в крови и повреждать развивающийся мозг
- Соблюдение низкобелковой диеты позволяет сохранять безопасный уровень ФА, что дает возможность нормально развиваться
- Употребление недопустимых продуктов не приведет к резкому ухудшению состояния ребенка, но вызовет проблемы в долгосрочной перспективе. Употребление продуктов, которые не входят в диету, не нужно рассматривать как «лакомство», поскольку это приведет к осложнениям больного ФКУ
- Ваш ребенок не перерастет ФКУ и должен соблюдать диету всю жизнь
Многие родители чувствуют необходимость поддержки от других семей с детьми, больными ФКУ, для того, чтобы удостовериться, что с их ребенком все будет в порядке. Вам, возможно, будет полезно пообщаться с родителями более старших детей с ФКУ о том, как они готовят пищу и что значит - жить на диете. Очень ободряют встречи с другими детьми, которые растут и хорошо развиваются.
Пока вы учитесь справляться с ФКУ, вы получите уверенность в будущем вашего ребенка. ФКУ станет частью вашей жизни, именно частью, а не всей жизнью!
Развитие
Развитие детей с ФКУ обычно четко совпадает с этапами физического и умственного развития здоровых детей; если наблюдаются какие-либо задержки, то они обычно не связаны с ФКУ. В возрасте примерно 18 месяцев познавательные, речевые и творческие способности большинства детей чрезвычайно развиваются. Они могут начать проявлять тревогу или страх относительно некоторых аспектов диеты и лечения. Очень важно говорить с ребенком о диете и лечении для того чтобы повысить понимание и избавиться от тревоги и страха.
Дети младше двух лет обычно не понимают какую пищу им можно есть, а какую нельзя, но они могут понять разницу между знакомой и не знакомой пищей. С раннего возраста, вы можете научить ребенка спрашивать вашего разрешения перед тем, как съесть что-то новое, или отказываться от пищи предложенной другими людьми.
Очень важно, чтобы братья и сестры тоже знали при диету при ФКУ. Вы можете привлекать старших детей для приготовления пищи и поощрять их помогать вам при кормлении младших детей с ФКУ, чтобы они знали продукты, которые относятся к диете при ФКУ. Когда старшие дети понимают необходимость специальной диеты для сохранения здоровья, их поведение и позитивное отношение может помочь ребенку с ФКУ принять свою диету и лечение.
Лечение и диета
Как показывает опыт, те дети, чьи родители вовремя обращаются за медицинской помощью, посещают регулярно больницу и регулярно сдают кровь, имеют лучший результат в борьбе с ФКУ. В больнице Вы получите информацию и знания, которые необходимы чтобы контролировать болезнь вашего ребенка. Сотрудничество с командой врачей поможет вам научиться жить с ФКУ и давать вашему ребенку лучший уход. Кормление ребенка с ФКУ более сложная задача, чем кормление ребенка без ФКУ. Вам помогут справляться с трудностями, связанными с кормлением и диетой. Соблюдение всех указаний очень важно и даст вам возможность обеспечить вашего ребенка соответствующим рационом, это поможет защитить его интеллектуальное развитие и физическое здоровье.
Кормление грудью и из бутылочки
Несмотря на то что у вашего ребенка ФКУ, он нуждается в некотором количестве ФА. Это количество ФА в рационе определяется уровнем ФА вашего ребенка. Как только достигнут безопасный уровень ФА, источником ФА становится грудное молоко или обычные детские смеси.
Вы можете выбрать грудное молоко в качестве источника ФА, и по-прежнему сохранять безопасный уровень ФА в крови вашего ребенка. Грудное молоко содержит гораздо меньше ФА, чем детские смеси. Однако, грудное молоко содержит слишком большое количество ФА, чтобы использовать только лишь его для питания ребенка с ФКУ, поэтому его нужно комбинировать со смесью без ФА. Вы, совместно с диетологом, можете решить будете ли вы сцеживаться и отмерять грудное молоко или вы будете прикладывать ребенка к груди. Если вы решите не использовать грудное молоко, обычная детская смесь является подходящей заменой молока для добавления ФА в рацион ребенка.
С младенчества и до двухлетнего возраста ребенка, у родителей, как правило, не возникает проблем с приятием питания ребенком. Врачи по ФКУ выпишут нужную смесь, которая в сочетании с грудным молоком или детской смесью, обеспечит вашего ребенка необходимым питанием. Количество лечебной смеси и грудного молока или детской смеси будут меняться время от времени, чтобы обеспечить необходимый и безопасный уровень ФА в крови. Для контроля потребления ФА очень полезно вести записи количества съеденной ребенком смеси и грудного молока. Вместе с диетологом вы определите, сколько вашему ребенку нужно грудного молока или детской смеси и сколько лечебной смеси.
Лечебная смесь
Лечебная смесь, грудное молоко или детская смесь - это единственная пища, которая необходима вашему ребенку до шести месяцев, любые дополнения, даже вода, могут привести к тому, что ваш ребенок не захочет есть смесь или молоко. Это особенно важно для детей с ФКУ, потому что для поддержания безопасных уровней ФА ему обязательно нужно употреблять определенное количество лечебной смеси и молока.
Один из главных способов контроля - точное измерение количества смеси с помощью весов с граммами.
Некоторые советы по приготовлению аминокислотной смеси
- Тщательно мойте и высушивайте руки перед тем как брать руками бутылочки и соски и перед тем как кормить ребенка
- Стерилизуйте бутылки и соски каждый раз перед использованием по крайней мере до шести месяцев
- Отмерьте аминокислотную смесь, добавьте необходимое количество воды и перемешайте, четко соблюдая инструкции, которые вы получили у врача по ФКУ
- Поместите готовую смесь, которую ребенок не съест немедленно, в самую холодную часть холодильника, обычно рядом с задней стенкой, сразу же после приготовления
- Когда вы выходите на улицу, кладите охлажденную смесь в термосумку со льдом
- Если ваш ребенок заболел и недостаточно ест, то нужно сообщить об этом врачу, чтобы откорректировать рацион, если это необходимо
Изменение смеси
Чтобы обеспечить пищевые потребности ребенка, аминокислотная смесь обычно меняется, когда ему исполняется год. Это изменение происходит постепенно, чтобы гарантировать принятие новой смеси ребенком.
Густая пища
Ребенок готов к употреблению густой пищи, когда он уже может сидеть с поддержкой и самостоятельно держать головку. В это время он может начать проявлять интерес к тому, что едите вы. Обычно это происходит в шесть месяцев. Даже когда ваш ребенок начал есть густую пищу, очень важно контролировать, чтобы он каждый день съедал необходимое количество предписанной лечебной смеси.
Как только ваш ребенок начинает употреблять густую пищу, вы должны считать потребление ФА при каждом приеме пищи. Ваш диетолог сообщит вам предписанное количество ФА. Используя справочное руководство по продуктам, вы сможете измерять количество продуктов и количество ФА, которое может съесть ваш ребенок.
Для контроля потребления лечебной смеси и густой пищи необходимо вести детализированную дневник рациона. Диетолог объяснит вам как заполнять дневник, который вы сможете приносить с собой в больницу или отправлять с анализами крови.
Введение густой пищи в рацион обычно занимает несколько месяцев, поскольку ребенок привыкает к новому вкусу и текстуре. Данная таблица дает некоторое представление о том когда ребенок может быть готов к новым видам пищи.
| Около 6 месяцев или когда ребенок может сидеть и держать головку | 8-9 месяцев | 10-12 месяцев | |
| Тип пищи | Вареная, размятая или процеженная пища | Мягкая, нарезанная пища | Пища, которую ребенок может есть самостоятельно, держа руками |
| Примеры |
|
|
|
Для того чтобы быть уверенными, что ваш ребенок укладывается в допустимые пределы потребления ФА в течение дня, вы можете использовать следующие методики:
- Решите сколько ФА ваш ребенок может съедать за каждым приемом пищи или перекусом
- Разделите общее количество ФА на каждый прием или перекус
- Выберите пищу, которую вы предложите ребенку, заранее, чтобы вы могли подсчитать запланированное потребление ФА каждый день
- Измерьте/ взвесьте количество пищи, которое обеспечит вашего ребенка необходимым уровнем ФА для данного приема пищи
- Сделайте запись того, сколько осталось не съеденным. Если ваш ребенок съел не все, вычитайте оставленное количество ФА из общего количества ФА, запланированного для данного приема пищи
- Предлагайте ребенку лечебную смесь в течение всего дня. Если ребенок уже съел всю лечебную смесь и все еще испытывает жажду, вы можете предложить воду или сок
Переход к питанию за столом
По достижению одного года ваш ребенок возможно уже есть фрукты и овощи, низко-белковые зерновые, каши, хлеб, макаронные изделия и крекеры. Ваш ребенок может проявлять интерес к еде, которую ест его семья, и обычно он готов к переходу от детского питания. Вовлекайте вашего ребенка в приемы пищи со всей семьей с раннего возраста.
Чтобы облегчить переход к питанию за столом используйте для приготовления пищи ребенку рецепты блюд с низким содержанием белка. Вы должны продолжать вести учет ФА - эти записи могут понадобится при визите к врачу или для анализов крови.
Как облегчить прием пищи
Отказ от приема пищи - обычное явления в раннем детстве, вне зависимости от того следует ли ребенок какой-либо диете или нет. Если родители смогут понять причины такого поведения ребенка, это поможет облегчить прием пищи.
Советы:
Сохраняйте позитивное отношение к диете вашего ребенка
Очень важно, чтобы семья и друзья имели позитивное отношение к диете ребенка. Позвольте ему сформировать собственное мнение о своей диете самостоятельно без негативного отношения с вашей стороны или со стороны других людей.
Предлагайте ребенку выбор, чтобы удовлетворить его растущую потребность в независимости
Как только дети начитают понимать, что они независимые люди, они могут выражать свои вкусы более явно. Вы можете предоставить ребенку на выбор два продукта или вовлекать в некоторые (безопасные) аспекты приготовления пищи. Ребенок также может захотеть есть самостоятельно. Позвольте ему использовать ложку или предложите на выбор продукты, которые можно есть руками.
Учтите, что ваш ребенок может и не быть голодным
После 12 месяцев дети уже не растут так быстро, это означает, что их аппетит может снизиться. Ребенок даст знать, когда он уже сыт. В этот момент нужно остановить кормление, даже если не все съедено. Аппетит ребенка может меняться в разные дни и ребенок будет есть, когда почувствует, что голоден.
Поддерживайте заведенный распорядок
Ребенок легко приучается жить по определенному распорядку. Он должен регулярно питаться, чтобы обеспечивать потребности растущего организма. Усаживайте ребенка за стол для приема пищи. Дети имеют короткую продолжительность концентрации внимания. Уделите 20-30 минут для основных приемов пищи и 10-15 минут для перекусов.
Мониторинг уровня ФА в крови
Цель - достичь диапазона ФА 2-6 мг/дл или 120-360 мкмоль/л
В этом возрасте, уровень ФА обычно измеряется при помощи анализа крови, взятой из пятки или большого пальца ноги, а когда ребенок подрастает - из пальца руки. Обычно в течение первых недель родителей обучают как брать кровь для анализа. Этому можно также обучить дедушек и бабушек и других близких.
Это процедура довольна проста, если у вас есть немного опыта. Важно донести до ребенка с помощью своих действий, что взятие крови для анализа обязательно и не подлежит обсуждению. Понимание важности этого анализа вашим ребенком, поможет ему в будущем контролировать ФКУ.
Вам предоставят специальную бумагу, необходимую для тестов и сообщат куда присылать карточки с анализом. Иногда кровь для анализа может браться из вены.
Советы родителям по забору крови для анализа:
- Берите кровь всегда в одно и то же время дня и при одинаковых условиях
- Помогите вашему ребенку понять чего стоит ожидать; ваши объяснения помогут уменьшить страх перед неизвестным.
- Поиграйте в теплой комнате или помойте ножки ребенка теплой водой для усиления кровоснабжения
- Сохраняйте непринужденную атмосферу. Спойте песенку или используйте те средства, которые успокаивают вашего ребенка
- Купите разноцветные пластыри с рисунками и позвольте ребенку выбрать
Во время первых недель лечения, пока устанавливается диета вашего ребенка, сдавать кровь необходимо один или два раза в неделю. После того как установлен регулярный распорядок приема пищи, частота взятия крови для анализа может быть сокращена до одного раза в неделю, но так как первые 12 месяцев - это время быстрого роста и смены диеты, иногда необходимо делать анализ чаще.
После 12 месяцев, частота анализов может быть сокращена. Детям старше года обычно рекомендуется делать анализ один раз в две недели. Дополнительные анализы могут понадобиться во время или после болезни для определения уровня ФА.
Перевод на русский язык – Королькова Надежда для Общества пациентов с фенилкетонурией, Москва
Оригинал статьи: National PKU alliance, www.npkua.org, http://www.npkua.org/pdf/PKU%20Binder%202011-Ch5.pdf
зачем берут анализ крови у новорожденного из пятки? О каких заболеваниях может сказать врачам и родителям малыша этот пяточный тест?
Зачем необходим скрининг новорожденного
Существует целый ряд довольно серьезных, как правило наследственных, заболеваний, наличие которых невозможно определить до рождения малыша, но которые крайне важно выявить, едва только младенец появился на свет. Потому что шансы на выздоровление ребенка резко повышаются в том случае, когда терапия начинает осуществляться еще до того, как проявились первые клинические симптомы. Именно для этого и проводится скрининг новорожденных. Обязательную часть которого составляет анализ крови из пятки младенца.
Анализ крови из пяточки младенца нередко еще называют «пяточный тест». Как правило, этот тест проводят на 4-й день жизни у доношенных ребятишек, и на 7-й день – у тех младенцев, кто родился раньше срока.
Не пугайтесь! Буквально пару капель крови, взятых у малыша из пятки, наносят на специальный бланк-фильтр – на этом вся процедура и заканчивается. Анализ проводится путем исследования сухого пятна крови – в медицине он известен как метод тандемной масс-спектрометрии. Пара минут относительного неудобства для новорожденного малыша, а также пара капель его крови для анализа могут раз и навсегда успокоить врачей и родителей новорожденного относительно того, какие тяжелые заболевания малышу уж точно не грозят. А именно…
Какие заболевания можно выявить с помощью скрининга новорожденных
В нашей стране с помощью анализа крови из пятки новорожденного проводят исследования на наличие как минимум пяти тяжелых наследственных заболеваний. Несмотря на то, что все эти «болячки» имеют наследственную природу, проявить себе каждая из этих болезней может в любом поколении – то есть у пораженного малыша может быть абсолютно здоровая семья, и только кто-то из дальних предков или дальних родственников может быть носителем той же болезни. Именно поэтому в роддоме тест делается всем малышам без исключения, независимо от того, известно ли о случаях тяжелых наследственных заболеваний в семье конкретного младенца или же нет.
Пять заболеваний, которые выявляются во всех российских роддомах с помощью анализа крови из пятки новорожденного (другими словами – путем скрининга новорожденного):
- Фенилкетонурия. В России это заболевание встречается примерно у одного из 10 тысяч малышей. Суть заболевания заключается в следующем: у ребенка отсутствует фермент, который расщепляет аминокислоту фенилаланин, входящую в состав большинства белковых продуктов. Не расщепляясь, эта аминокислота и ее производные накапливается в организме и к определенному моменту становятся крайне токсичными – они поражают нервную систему и в первую очередь мозг младенца. Если финилкетонурию не лечить (особенно на ранних стадиях), то это приводит в 100% случаев к умственной отсталости ребенка. При адекватном и раннем лечении (прием специальных препаратов и строгая диета) малыш будет развиваться абсолютно нормально, без задержек, но останется на всю жизнь носителем заболевания.
- Врожденный гипотиреоз. Заболевание характеризуется недостаточной выработкой некоторых гормонов. Без должного лечения у малыша наблюдаются задержки в физическом и умственном развитии – например, позже формируются кости и суставы, позже режутся первые зубки, медленно развиваются нервные ткани и мозг. В итоге ребенок развивается с серьезными физическими и интеллектуальными дефектами. Если же болезнь диагностируется еще до появления клинических симптомов (то есть еще в период новорожденности), назначается гормональная терапия. Которая дает возможность полностью пресечь развитие болезни. В России врожденный гипотиреоз встречается у одного новорожденного на 5 тысяч.
- Галактоземия. В основе этого наследственного заболевания лежит серьезное нарушение обмена веществ – ген, который отвечает за преобразование галактозы (вещества, которое содержится в молоке) в глюкозу, мутирован. Галактоза не подвергается ферментации, а накапливается в организме, оказывая сильное токсическое воздействие на центральную нервную систему. Кроме того, сильно страдают печень и зрение малыша. Если заболевание игнорировать, то ребенку грозит слепота, хроническое поражение печени и умственная отсталость в развитии. В теории галактоземия приводит к летальному исходу. Но при своевременном лечении специальной диетой, полностью исключающей из рациона малыша молоко и молочные продукты, ребенок имеет все шансы полноценно расти и развиваться. Галактоземия встречается примерно у одного новорожденного на 20 тысяч малышей.
- Адреногенитальный синдром. Тяжелое заболевание характеризуется нарушением выработки гормонов корой надпочечников. В организме образуется избыток половых гормонов и глюкокортикоидов. Что вызывает резкое нарушение солевого обмена, нарушение роста (зачастую после 12 лет рост прекращается вовсе) и неправильное развитие половых органов. Девочки, как правило, развиваются «по мужскому типу». Чтобы избежать развитие болезни, малышам назначают гормональную терапию. В России адреногенитальный синдром встречается у одного новорожденного на 5,5 тысяч малышей.
- Муковисцидоз (кистозный фиброз). Это системное наследственное заболевание, которое характеризуется тяжелейшими нарушениями в органах дыхания и органах пищеварения. Причина заболевания – генная мутация. В России муковисцидоз встречается у одного малыша на 10 тысяч новорожденных. Терапия, которую назначают при ранней диагностике, способна резко сократить проявления болезни.
Что происходит после теста?
В последние годы в некоторых регионах страны список заболеваний, анализ на которых включен в программу обязательного скрининга новорожденного, значительно расширился. Формируется этот список на основании рекомендаций Всемирной Организации Здравоохранения (ВОЗ).
Сама процедура считается абсолютно безопасной для младенца, а результаты она дает весьма полезные – выявление наследственных болезней на столь ранней стадии позволяет подобрать адекватную стратегию лечения и избежать инвалидности у больных детей в будущем.
Анализ крови из пяточки новорожденного, взятый в роддоме, передается в лабораторию. А оттуда результаты, спустя максимум 10 дней, поступают в общую базу и непосредственно в ту поликлинику, где далее наблюдается малыш.
Чаще всего результаты скрининга новорожденного оказываются отрицательными – факт об этом заносится в медицинскую карту ребенка, и дело с концом. Как правило, при отрицательном ответе на анализ из пятки новорожденного, с родителями никто это не обсуждает. Если же тест показал положительный результат на какое-либо из заболеваний – с родителями или опекунами младенца мгновенно связываются медики. Однако это еще не приговор! Родителям сообщают о том, что у ребенка выявлено лишь подозрение на наследственную болезнь и назначают дату ретестирования. Очень важно провести повторный анализ как можно скорее.
И только в том случае, если повторное исследование в медико-генетическом центре дало положительный результат, малышу назначается соответствующая терапия.
Все болезни, наличие которых способен выявить неонатальный скрининг – тяжелые и приводят к глубокой инвалидности в будущем. Однако, при ранней диагностике – еще на стадии новорожденности ребенка (первые 28 дней жизни младенца) – эти недуги можно либо вылечить полностью, либо жестко приостановить их развитие на той стадии, когда болезнь еще не приобрела необратимые формы.
Именно поэтому крошечных деток не только можно, но и обязательно нужно подвергать анализу крови из пяточки спустя несколько дней после рождения. Обычно это делают в роддоме. Но если вы планируете домашние роды, либо собираетесь покинуть роддом спустя уже день-два после родов, обязательно уточните у своего врача, где и как вы сможете провести скрининг новорожденного.