Миотонический синдром что это такое
Миотонический синдром, миотония у детей и взрослых: причины, симптомы, лечение
Миотонический синдром (миотония) — группа симптомов, возникающих при нарушении мышечного тонуса и дисфункции мышечных волокон. Они слабеют в состоянии покоя и спазмируются при чрезмерном напряжении. Сжатые мышцы не могут полностью расслабиться сразу после сокращения. Миотония является проявлением мышечных нарушений, при которых они теряют способность к релаксации после сокращения. Временные расстройства проявляются скованностью движений. Наиболее частыми проявлениями патологии являются миотонические атаки.
Термин «миотония» в переводе с латинского языка означает «мио» — мышца и «тонус» — напряжение. Заболеванию подвержены только скелетные мышцы, обеспечивающие произвольные движения, в том числе мышцы головы, лица и языка. Гладкая мускулатура внутренних органов сокращается непроизвольно и не страдает от данного недуга.
Это заболевание нервно-мышечной системы проявляется мышечной гипотонией и слабостью, которая быстро сменяется спазмами, болью и напряженностью. В ответ на внешние раздражители мышцы всегда сокращаются, но не сразу расслабляются. Для них этот процесс сложный и трудоемкий.
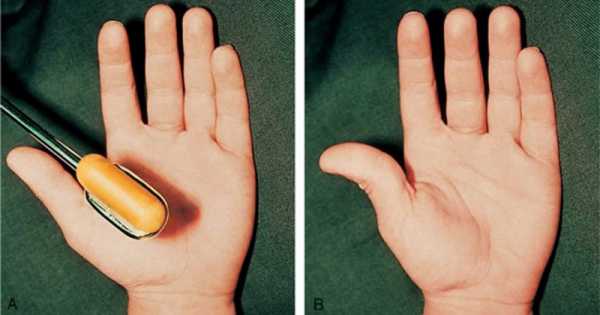
мышечная реакция на простукивания у больного миотонией
Мышечная слабость — причина неправильной осанки у детей, невозможности держать ровно спину и правильно говорить. Больные дети начинают сидеть и ходить позже своих здоровых сверстников, у них нарушается работа суставов. Поражение мышц передней брюшной стенки приводит к нарушению работы ЖКТ и желчевыводящих путей, развитию хронических запоров. Слабый мышечный тонус может спровоцировать недержание мочи, близорукость, искривление позвоночника, остеохондроз. Школьники с миотоническим синдромом быстро устают даже после незначительных психофизических нагрузок, процесс обучения дается им с большим трудом. Их часто мучают интенсивные головные боли, что приводит к нарушению психики и снижению трудоспособности.
- Миотония — наследственное заболевание, связанное с поражением различных отделов головного мозга. Эта геномная патология может развиться у любого человека независимо от пола, возраста и национальности. Проявляется она у грудничков, детей постарше, подростков и взрослых людей. Миотонические атаки длятся от нескольких секунд до минут, а их интенсивность колеблется от дискомфортных ощущений до болезненных дисфункциональных нарушений. Спровоцировать миотоническую атаку могут следующие факторы: физическое перенапряжение, длительный отдых, холодовое воздействие, резкие и громкие звуки.
- Приобретенная форма миотонического синдрома связана с травмами, полученными во время родов, метаболическими нарушениями, перенесенным рахитом. У грудных детей с трудом разжимаются пальцы рук при захвате предмета, слабо выражен сосательный рефлекс. Дети постарше мало играют в подвижные игры, долго поднимаются после падения, сторонятся сверстников.
Лечение патологии зависит от причин, спровоцировавшей ее появление и развитие.
Этиология
Этиологические и патогенетические факторы миотонического синдрома точно не определены. Ученые выделяют две формы патологии — врожденную и приобретенную.
- В основе недуга могут лежать генетические нарушения. Синдром передается по наследству детям от больных родителей. Чтобы болезнь проявила себя клинически, необходимо получить от матери и отца по одной измененной копии гена. В этом случае мутантный ген подавляет его нормальную копию, и происходит развитие патологии. Если ребенок наследует мутантный ген только от одного из родителей, он становится его носителем, но не болеет. При передаче здоровой копии гена ребенок рождается абсолютно здоровым. Врожденная форма патологии имеет различные проявления.
- Спровоцировать развитие патологии могут другие нервно-мышечные и эндокринные заболевания, травмы при родах, постгипоксическая энцефалопатия новорожденных, метаболические расстройства, гиподинамия беременной женщины в течение всего периода вынашивания ребенка. Приобретенный миотонический синдром хорошо поддается лечению, в отличие от генетического типа недуга.
Механизм развития миотонии одинаков у всех больных. Ослабленные мышцы под воздействием определенных факторов сильно тонизируются и даже парализуются. Наступает так называемая миотоническая атака. Она развивается при попытке больного сделать движение, которое требует участия пораженных мышц. К таким провоцирующим факторам относятся: холод, стресс, эмоциональный всплеск, громкий звук, длительное неподвижное положение.
Симптоматика
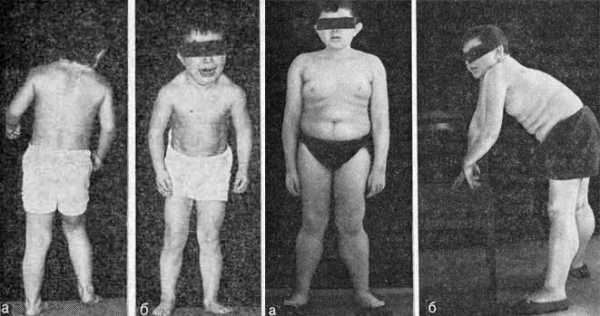
Клинические признаки патологии зависят от локализации очага поражения. При нарушении активности мышц ног, рук, плеч, шеи, лица у больных возникают трудности в процессе ходьбы, удержания осанки, разговора, управления мимикой, приема пищи.
К общим проявлениям синдрома относятся:
- мышечная слабость,
- быстрая утомляемость,
- апатия,
- нарушение работы ЖКТ, хронические запоры и кишечные колики,
- энурез,
- цефалгия невыясненного происхождения,
- сутулость, сгорбленность,
- миопия,
- речевые нарушения,
- нарушение равновесия, потеря устойчивости,
- неуверенная походка при крутом подъеме и резком спуске,
- снижение интеллекта.
Впервые симптомы патологии обнаруживают у детей раннего возраста. Больные малыши позже здоровых начинают держать головку, ползать, ходить и говорить. Причем эти действия даются им с большим трудом и часто только с помощью взрослых. Их тело плохо поддается управлению, оно как будто не слушает своего хозяина. Малыш, пытаясь встать, замирает и падает. Затем ребенок поднимается и после приложения определенных усилий выполняет задуманное действие. Пытаясь тронуться с места, малыш снова падает, словно парализованный, затем поднимается и движется более уверенно. Чем больше попыток он совершает, тем более правильными становятся его движения. При выполнении нескольких последовательных сокращений мышца разрабатывается, и ее дальнейшее расслабление происходит легче.
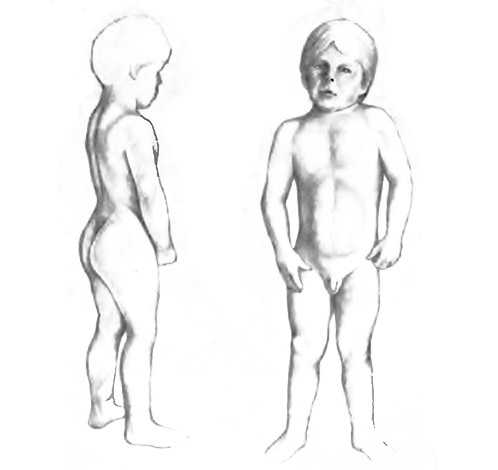
ребенок с врожденной миотонией: повышенный тонус мышц плечевого пояса, гипертрофия мышц бедра
У больных нарушается стул, возникает хронический запор, развивается дисфункция мускулатуры мочевого пузыря и желчевыводящих протоков. При поражении мышечных волокон органа зрения появляются симптомы миопии. Больные дети плохо говорят, быстро устают, часто жалуются на головную боль, теряют равновесие при подъеме по лестнице или во время быстрой ходьбы. Спровоцировать спазм пораженных мышц ног и боль может легкое постукивание по икрам. Это один из диагностических критериев синдрома, который можно идентифицировать самостоятельно. Болезненный спазм сопровождается появлением мышечных валиков на ноге, которые сохраняются в течение нескольких минут после расслабления.
У взрослых перенапряженные мышцы гипертрофируются, становятся крупными, накачанными. Такие люди выглядит как качки или бодибилдеры.
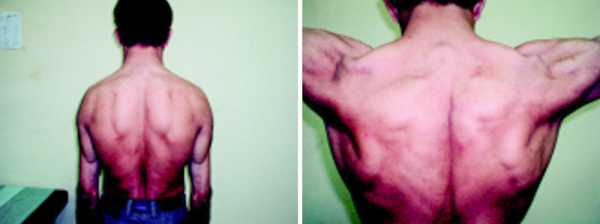
мышечная гипертрофия при миотонии у взрослых
При поражении лицевых и шейных мышц страдает внешний вид больных. Миотонические спазмы изменяют тембр голоса, нарушают глотательные и дыхательные процессы, что проявляется одышкой и дисфагией. Больные без проблем ходят по ровной поверхности, но с трудом поднимаются по ступенькам, особенно преодолевая первый пролет. Феномен приседания заключается в невозможности полностью присесть, чтобы стопа от носка до пятки стояла на полу. Больные не держат равновесие и сразу падают.
миоплегияДиагностика
Лечебно-диагностическими процедурами при миотоническом синдроме занимаются специалисты в области неврологии, логопедии, ортопедии, терапии, педиатрии и офтальмологии.
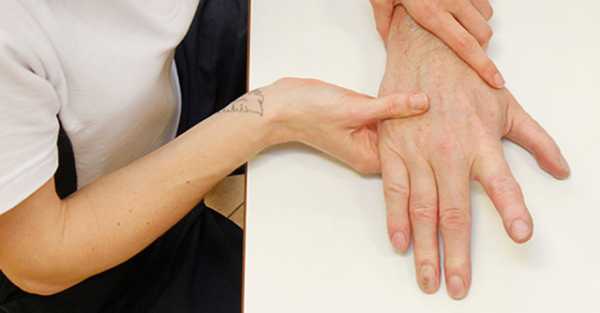
Диагностические мероприятия:
- Сбор наследственного анамнеза — выяснение сведений о наличии аналогичного синдрома у ближайших родственников.
- Простукивание мышц перкуссионным молоточком с целью обнаружения дефекта.
- Специфический тест для выявления миотонической реакции на раздражители – нагрузку, холод, резкий звук. Сначала у больных появляются миотонические разряды, а затем исчезают все характерные феномены. Больным предлагают быстро разжать кулак, открыть зажмуренные глаза, встать со стула. Для выполнения этих простых действия пациентам необходимы время и определенные усилия.
- Электромиография проводится с целью исследования биоэлектрических потенциалов, возникающих в скелетных мышцах человека при возбуждении мышечных волокон. При миотонии регистрируются миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика».
- Анализ крови на параклинические и биохимические показатели.
- Молекулярно-генетическое исследование.
- Биопсия и гистологическое исследование мышечных волокон.
Лечебные мероприятия
Лечение миотонического синдрома зависит от его причин. Если недуг приобретенный, врачи назначают комплексную терапию, устраняющую этиопатогенетические факторы и последствия их воздействия. Врожденный синдром — неизлечимое заболевание, которое остается с ребенком на всю жизнь. Общетерапевтические мероприятия способны существенно облегчить состояние больных, поэтому являются обязательно необходимыми.
Всем больным проводят симптоматическую терапию, направленную на нормализацию мышечного тонуса, их быстрое восстановление после перенапряжения или сокращения, стабилизацию метаболизма в организме. Обычно назначают ноотропные, сосудистые, нейропротекторные и метаболические средства:
- «Актовегин» активизирует обмен веществ в тканях, улучшает трофику и метаболизм на клеточном уровне, стимулирует процесс регенерации.
- «Пантогам» – ноотропный препарат, запускающий метаболические процессы в клетках мозга и устраняющий последствия психомоторного возбуждения.
- «Фенитоин» – миорелаксант с противоконвульсивным действием.
- «Элькар» повышает активность мозга и нормализует обменные процессы.
- Диуретики «Верошпирон» и «Гипотиазид» применяют для снижения калия в крови.
- «Церебролизин» стимулирует мозговую деятельность, проявляя функциональную нейромодуляцию и нейротрофическую активность.
- «Кортексин» – мощный антиоксидант и антигипоксант, улучшающий функции головного мозга и повышающий устойчивость организма к внешним раздражителям.
- В тяжелых случаях проводится иммуносупрессивная терапия – внутривенное введение иммуноглобулина человека, «Преднизолона», «Циклофосфамида».
- Кроме медикаментозного лечения необходимо непосредственное воздействие на мышцы с целью их тренировки и укрепления. Для этого проводят несколько курсов массажа, занятия ЛФК, закаливание. Ежедневная гимнастика и сеансы массажа помогут восстановить мышечную активность, нормализовать тонус мышц, устранить неприятные симптомы синдрома.
- Для улучшения самочувствия необходимо придерживаться диетического питания с ограничением солей калия.
- Взрослым пациентам полезно плавание в бассейне, а малышам – в домашней ванне.
- Физиотерапевтические процедуры, эффективные при миотонии — озокеритовае аппликации, электрофоретическое воздействие, иглорефлексотерапия, электростимуляция.
- Коррекционные занятия с больными детьми в специализированном медицинском центре.
- Занятия с логопедом, психологом, дефектологом.
Специалисты рекомендуют своим пациентам избегать воздействия провоцирующих факторов: конфликтных ситуаций, психоэмоциональных всплесков, физического перенапряжения, долгого пребывания в одной и той же позе, переохлаждения.

Если родители приложат определенные усилия и начнут лечить детей с раннего детства, они вырастут и будут жить полноценной жизнью. Сразу после того, как родится малыш, ему следует организовать правильный уход: кормить грудью, часто и подолгу гулять с ним на свежем воздухе, делать массаж, выполнять зарядку по утрам. Эти профилактические мероприятия помогут избежать дальнейшего прогрессирования мышечной слабости.
Если не предавать особого значения симптомам патологии, можно нанести непоправимый вред здоровью малыша. Он начнет позже сверстников держать головку, сидеть и ходить, в дальнейшем возникнут проблемы с речью. К тяжелым осложнениям синдрома относятся: постоянная мигрень, энурез, снижение остроты зрения, искривление позвоночника.
Медико-генетическое консультирование показано всем лицам, планирующим беременность и находящимся в группе риска. Это касается семейных пар, у которых в роду имелись хромосомные аномалии. Женщинам во время беременности показана пренатальная диагностика генетических расстройств, которая включает неинвазивные и инвазивные методики. Для этого проводят УЗИ плода, скрининг биохимических сывороточных маркеров, амниоцентез, плацентоцентез, биопсию ворсин хориона, кордоцентез.
Миотонический синдром не так страшен, как о нем думают обеспокоенные молодые мамочки. Своевременная и корректная диагностика, а также адекватное лечение позволяют таким малышам вырасти здоровыми и счастливыми.
Видео: лекция о врожденном миотоническом синдроме
Видео: миотоническая дистрофия
Миотонический синдром - лечение, симптомы, причины, диагностика
Миотония это не заболевание, а симптом группы мышечных нарушений, для которых характерно нарушение способности мышцы к релаксации после сокращения. У большинства людей миотоническая атака имеет временный характер и проявляется выраженной скованностью, которая возникает после выполнения определенного вида движений. Невозможность сжатых мышц расслабиться и трудности при вставании из сидячего положения являются наиболее частыми проявлениями миотонической атаки.
Длительность миотонической атаки может быть продолжительностью от секунд до минут и по интенсивности колебаться от небольшого дискомфорта до выраженного нарушения двигательных возможностей. Как правило, миотоническая атака возникает после интенсивной физической нагрузки или после длительного периода отдыха, но иногда возникает как реакция на низкие температуры или даже резкий звук.
При миотонии идет вовлечение только скелетной мускулатуры, выполняющей произвольные движения. Таким образом, миотоническая атака может возникнуть только при выполнении осознанных движений, но не поражает мышцу сердца или мышечные структуры пищеварительного тракта. В зависимости от типа и серьезности миотония может возникнуть в ногах, лице, руках, в плечах, стопе, в мышцах век глаз и даже способности говорить четко.
Как правило, миотонические синдромы являются генетически детерминированными и могут быть у любого человека. Миотонические нарушения могут быть как у женщин, так и мужчин и в любом возрасте могут быть симптомы миотонических атак. В зависимости от выраженности миотонических симптомов первые признаки этих нарушений могут появиться в подростковом возрасте или у взрослых. Наследование миотонических синдромов происходит двумя путями аутосомально-доминантным генетическим паттерном или аутосомально-рецессивным генетическим паттерном.
Аутосомально доминантное наследование
Нарушения, связанные с аутосомально-доминантным наследованием обычно проявляется в каждом поколении без исключений. Аутосомальный означает то, что генетическая ошибка может располагаться на любой хромосоме, в каждой клетке организма, за исключением половых хромосом. Доминантный означает, что достаточно одного родителя носителя дефектного гена для передачи заболевания по наследству. И так как нарушения могут передаваться любым родителем, то любой ребенок от родителя с генетическим дефектом имеет 50% шанс наследовать это нарушение. Выраженность нарушения и возраст дебюта могут варьировать в зависимости от конкретного индивидуума.
Аутосомально-рецессивное наследование
При аутосомально-рецессивном наследовании заболевание появляется в одном поколении и обычно без наличия семейного анамнеза этого состояния. Оба родителя могут быть носителями этого гена. Носитель дефектного гена может не отмечать какой-либо симптоматики. Рецессивность означает то, что для проявления этого гена и заболевания необходимо, чтобы у обоих родителей был дефектный ген. Дети обоего пола могут наследовать дефектный ген по этому типу наследования. При каждой беременности у 25 % детей есть вероятность наследования дефектного гена от обоих родителей. Если ребенок наследует дефектный ген от одного родителя, то он будет носителем дефектного гена, но, скорее всего, проявляться это симптомами не будет. В 50% случаев ребенок будет носителем гена. В то же время вероятность того, что ребенок не наследует дефектный ген и не будет носителем и не будет иметь клиническую манифестацию, составляет 25%.
Виды миотоний
Миотония конгенита
Миотония конгенита наиболее часто встречающаяся непрогрессирующая форма миотонического синдрома и вызвана мутацией гена, отвечающего за натриевые ионные каналы мышц. Эта форма миотонии не оказывает влияние на продолжительность жизни и имеет незначительное влияние на структуру тела или рост опорно-двигательного аппарата. Существует две формы миотонии конгенита в зависимости от типа наследования.
Частой и довольно тяжелым типом миотонии конгенита является генерализованная миотония Бейкера, и наследование этого заболевания происходит по аутосомально-рецессивному типу. Дебют этой формы миотонии бывает в детстве или раннем подростковом возрасте, но иногда при тяжелом течение дебют может быть и в раннем детском возрасте. Симптоматика может нарастать прогрессивно, в течение нескольких лет после постановки диагноза или постепенно нарастать до достижения пациентом двадцатилетнего возраста.
Аутосомально - доминантная форма наследования называют болезнью Томсена. Заболевание названо в честь датского врача Асмуса Юлиуса Томсена у которого было это заболевание и который проследил наличие этого заболевания в своей семье. Симптомы миотонии Томсена, как правило, значительно мягче, чем при миотонии Бейкера и хотя дебют заболевания бывает раньше, и первые признаки становятся заметными в раннем детстве и иногда при рождении. В редких случаях симптомы могут быть незначительными, в течение многих лет после постановки диагноза.
Основным симптомом обеих заболеваний является генерализованная миотония, вызванная произвольными движениями. Как правило, такая симптоматика провоцируется значительной физической нагрузкой или наоборот длительным периодом отдыха и мышечной релаксации. Миотония более выражена в ногах, что вызывает затруднения при ходьбе и иногда даже падения. Также миотонией бывают охвачены мышцы плечевого пояса и головы, что может вызвать затруднения при хватании предметов, жевании или мигании. В редких случаях при миотонии Бейкера может быть обездвиживающая слабость, появляющаяся после миотонической атаки.
После миотонической атаки при обоих типах миотонии снять скованность можно с помощью повторных движений в скованных мышцах. Как правило, скованность увеличивается после нескольких первых сокращений заинтересованных мышц, а затем после пяти сокращений миотоническая скованность исчезает, что позволяет восстановить нормальную работу мышц на определенный промежуток времени. Это эффект называется эффектом разминки (разогрева) и позволяет людям с миотонией заниматься тяжелыми физическими нагрузками и силовыми видами спорта.
И хотя миотония конгенита не оказывает значительного влияния формирование опорно-двигательного аппарата, в то же время миотония влияет на размер определенных мышц. Обе миотонии Бейкера и Томсена могут вызывать необычное увеличение размеров скелетных мышц, особенно в области ног ягодиц, но также и в области рук плеч и мышц спины. Такое увеличение может считаться мышечной гипертрофией и иногда такие пациенты выглядят как настоящие атлеты. При миотонии Бейкера гипертрофия мышц более выражена, чем при миотонии Томсена.
Парамиотония конгенита
Эта миотония является редкой патологией, связанной нарушением натриевых каналов и передающаяся по аутосомно-доминантному типу. Этот тип миотонии не сокращает продолжительность жизни, и интенсивность миотонии сохраняется стабильной в течение жизни. Дебют заболевания возникает в период между рождением и ранним детским возрастом. Характерным симптомом парамиотонии конгенита является генерализованная миотоническая скованность, которая в большей степени поражает руки и лицо, а также области шеи и руки. Так же, как и при других непрогрессирующих миотониях, парамиотония конгенита провоцируется интенсивными произвольными нагрузками, а в некоторых случаях провоцируются низкой температурой. Во многих случаях, миотонические атаки индуцированные холодом и связанную с ними мышечную скованность можно снять теплом.
Атаки миотонической скованности часто сопровождаются обездвиживающей слабостью в заинтересованных зонах. Слабость может быть более длительной, чем эпизод миотонической скованности, ослабляя мышцы на период от нескольких минут до нескольких часов. Но слабость не характерна для парамиотонии конгенита. Кроме того, в отличие от других непрогрессирующих форм миотонии, для парамиотонии не характерно наличие эффекта разминки, при котором происходит снижение скованности после нескольких сокращений мышц. Наоборот, миотоническая скованность обычно увеличивается после продолжающейся активности мышц, что еще больше снижает двигательную активность. Этот феномен обычно называют парадоксальной миотонией.
Синдром Шварц Джемпела
Синдром Шварц - Джемпела является наиболее тяжелой формой непрогрессирующей миотонии. Это очень редкий тип миотонии передается по аутосомально-рецессивному типу. Дебют заболевания возникает или сразу после рождения или через небольшое время после рождения. Характер и интенсивность симптоматики могут иметь индивидуальные особенности. Одним из главных симптомов является миотоническая скованность, наиболее выраженная в лице и бедрах. При этой миотонии имеется тенденция к падениям нарушениям речи и лицевым изменениям. Как и при миотонии конгенита дополнительные сокращения мышц вызывают эффект разминки и снижают скованность. Но эффект разминки имеет незначительную степень, а некоторых случаях отсутствует. Для синдрома Шварца-Джемпела характерны различные скелетные деформации. Эти деформации к проблема роста организма и, как правило, это проявляется уменьшением роста, а также аномалиям лица, что придает лицу маскообразную форму. Другие симптомы включают гипертрофированные бедра, атрофированные плечевой пояс и длительные мышечные подергивания, а иногда и нарушения интеллектуальной сферы.
Причина синдрома Шварца-Джемпела не известна. Не исключается, что этот вид миотонии является разновидностью мышечных нарушений, а также возможно иметь связь с нарушениями в нервной системе или является сочетанием нарушениям в мышцах и нервах.
Диагностика
Даже на основании физикального обследования врач может провести определенные диагностические тесты, такие как миотоническая реакция на нагрузку, воздействие холодом или другие стимуляции. Но очень важно диффенцировать эти миотонии с прогрессирующими заболеваниями, такими как миотонические дистрофии. И для дифференциального диагноза необходимы специальные методы исследования. Эти исследования включают проведение ЭМГ, который позволяет определить электрическую активность мышечных ткани; лабораторные исследования (анализы крови и анализ ДНК) биопсию мышц, которая бывает иногда необходима для окончательного диагноза мышечного заболевания.
Лечение
В настоящее время не существует методов лечения, позволяющих избавиться полностью от какого либо миотонического синдрома. Лечение носит симптоматический характер. Если миотонические атаки становятся интенсивными, возникает необходимость применения медикаментов, позволяющих снизить симптоматику. Наиболее известным препаратом является мексилитен, а также такие препараты как гуанин, прокаинамид, тегретол, фенитоин. Но все эти препараты имеют массу побочных эффектов и поэтому их длительное употребление не желательно. Наиболее оптимально, когда пациент знает провоцирующие миотонические атаки факторы и старается по возможности избегать провоцирующих ситуаций, а после атак дает возможность мышцам восстановиться с помощью отдыха.
Миотонический синдром у взрослых - что это, как лечить миотонический болевой синдром у женщин, диагностика и лечение миотонии в Москве
Содержание↓[показать]Миотонический синдром - это комплекс нервно-мышечных нарушений, при котором у человека нарушается способность мышцы расслабляться после сокращения. Для миотонического синдрома характерен преимущественно наследственный характер – это геномная патология аутосомно-рецессивного или аутосомно-доминантного типа наследования. Характерными симптомами миотонии являются: мышечная гипотония и слабость, впоследствии – спазмы, боли и напряженность.
Диагностика и лечение миотонического синдрома проводится в центре реабилитации Юсуповской больницы – ведущем медицинском центре Москвы, оснащенном новейшим медицинским оборудованием, позволяющим нашим высококвалифицированным специалистам точно и в короткие сроки обнаружить патологию и подобрать наиболее эффективную индивидуальную схему лечения.

Формы заболевания
При отсутствии лечения даже умеренный миотонический синдром может привести к нарушению осанки, нарушению работы суставов, нарушению деятельности желудочно-кишечного тракта, желчевыводящих путей, хроническим запорам (вследствие поражения мышц передней брюшной стенки).
Ослабление мышечного тонуса может сопровождаться недержанием мочи, развитием близорукости, искривлением позвоночника, остеохондрозом. У больных с миотоническим синдромом отмечается появление интенсивных головных болей, вследствие чего нарушается психика и снижается трудоспособность.
Миотония может быть врожденной - иметь генетический характер. В этом случае появление геномной патологии связано с поражением того или иного отдела головного мозга. Миотонический синдром может наблюдаться у людей любого возраста и пола. Продолжительность миотонической атаки составляет несколько секунд или минут. Различна и интенсивность атаки – у одних больных могут возникать дискомфортные ощущения, у других - болезненные дисфункциональные нарушения. Развитие миотонической атаки может быть обусловлено физическим перенапряжением, длительным отдыхом, холодовым воздействием, резкими и громкими звуками.
Существует и приобретенная форма миотонии. Она возникает при травмах, полученных в процессе родов, метаболических нарушениях, после перенесенного рахита. Грудные дети не могут разжать пальцы рук, пытаясь захватить предмет, у них отмечается слабая выраженность сосательного рефлекса. У детей постарше возникают трудности с подъемом тела после падений, подвижными играми.
Для выбора грамотного метода лечения миотонического синдрома специалисты Юсуповской больницы проводят предварительное комплексное обследование больных, позволяющее выявить причину, которая спровоцировала развитие данной патологии.
Как проявляется у женщин и мужчин?
Клиническая картина миотонии зависит от того, где локализован очаг поражения. У больных с нарушением активности мышц рук, ног, лица, плеч, шеи, отмечаются сложности с ходьбой, удержанием осанки, речевой функцией, управлением мимикой, приемом пищи.
К общим симптомам миотонического синдрома относят:
- мышечную слабость;
- апатию;
- быструю утомляемость;
- энурез;
- нарушение функции желудочно-кишечного тракта: кишечные колики и хронические запоры;
- цефалгию невыясненной этиологии;
- сгорбленность, сутулость;
- потеря устойчивости; нарушение равновесия;
- миопию;
- нарушения речи;
- снижение интеллекта;
- неуверенную походку при резком спуске и крутом подъеме.
Чаще всего болезнь впервые проявляется в детском возрасте. Больным малышам сложнее, чем здоровым детям, держать голову, они позже начинают ходить, разговаривать. Ребенок с миотоническим синдромом не в состоянии нормально контролировать свои движения.
У них отмечается нарушение стула, развитие хронических запоров, дисфункции желчевыводящих протоков и мочевого пузыря.
Миотонический синдром у взрослых может проявляться гипертрофией перенапряженных мышц, увеличением их объема. Больные с миотоническим синдромом часто выглядят как бодибилдеры.
При поражении мышц лица и шеи (шейный миотонический синдром) наблюдается изменение внешнего вида больного, изменение тембра голоса, нарушение глотательных и дыхательных процессов – развивается одышка и дисфагия.
Как обнаружить у взрослых?
Для того, чтобы выявить миотонический болевой синдром, специалисты Юсуповской больницы используют мультидисциплинарный подход с привлечением неврологов, ортопедов, терапевтов, офтальмологов.
В первую очередь врач собирает наследственный анамнез пациента: выясняет сведения о наличии миотонического синдрома у членов семьи больного. Затем с помощью перкуссионного молоточка простукивает мышцы для выявления дефекта.
Для того, чтобы выявить миотоническую реакцию на внешние раздражители (нагрузки, резкие звуки, холод) специалисты Юсуповской больницы проводят специфический тест.
Исследовать биоэлектрические потенциалы, возникающие в скелетных мышцах при возбуждении мышечных волокон, позволяет электромиография.
Для определения параклинических и биохимических показателей проводится анализ крови.
Дополнительно назначается проведение молекулярно-генетического исследования, биопсии и гистологического исследования мышечных волокон.
Как вылечить?
Метод лечения миотонического синдрома напрямую зависит от причины, спровоцировавшей его возникновение.
Больным с приобретенным миотоническим синдромом назначается проведение комплексной терапии, направленной на устранение этиопатогенетических факторов и последствий их воздействия.
Врожденный миотонический синдром считается неизлечимым заболеванием. Для облегчения состояния больных необходимо проведение общетерапевтических мероприятий.
Всем больным назначается симптоматическая терапия, позволяющая нормализовать мышечный тонус, стабилизировать метаболизм в организме, способствующая быстрому восстановлению мышц после сокращения и перенапряжения. Чаще всего рекомендуется прием сосудистых, ноотропных, метаболических и нейропротекторных средств:
- актовегина – для активации обмена веществ в тканях, улучшения трофики и клеточного метаболизма, стимуляции процесса регенерации;
- пантогама – ноотропного препарата для запуска метаболических процессов в клетках мозга и устранения последствий психомоторного возбуждения;
- фенитоина – миорелаксанта, оказывающего противоконвульсивное действие;
- элькара – для повышения активности головного мозга, нормализации обменных процессов;
- кортексина – мощного антиоксиданта и антигипоксанта, используемого для улучшения функций головного мозга и повышения устойчивости организма к внешним раздражителям;
- верошпирона, гипотиазида – диуретиков, снижающих уровень калия в крови;
- церебролизина – для стимуляции мозговой деятельности благодаря функциональной нейромодуляции и нейротрофической активности.
Тяжелые случаи требуют применения иммуносупрессивной терапии – внутривенного введения иммуноглобулина, преднизолона, циклофосфамида.
Кроме медикаментозной терапии в клинике реабилитации Юсуповской больницы проводятся лечебные мероприятия, непосредственно воздействующие на мышцы и направленные на их тренировку и укрепление: массаж, лечебная физкультура, закаливание. Ежедневное выполнение гимнастики и сеансов массажа способствует восстановлению мышечной активности, нормализации тонуса мышц, устранению неприятных симптомов миотонического синдрома.
Для лечения миотонического синдрома в клинике реабилитации Юсуповской больницы пациентам назначается проведение физиотерапевтических процедур: озокеритовых аппликаций, электрофоретического воздействия, иглорефлексотерапии, электростимуляции.
Для улучшения самочувствия больных в Юсуповской больнице организовано диетическое питание с низким содержанием солей калия.
В целях профилактики возникновения миотонических атак пациентам рекомендуется по возможности стараться исключить провоцирующие факторы патологии: физическое перенапряжение, конфликтные ситуации, психоэмоциональные всплески, переохлаждение, долгое пребывание в одной и той же позе.
Записаться на прием к специалисту центра реабилитации Юсуповской больницы, получить подробную информацию о методах лечения миотонического синдрома и их стоимости можно по телефону или онлайн на сайте Юсуповской больницы.
Автор
Константин Юрьевич КазанцевВрач - невролог, ведущий специалист отделения неврологии
Список литературы
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Бадалян Л. О. Невропатология. — М.: Просвещение, 1982. — С.307—308.
- Боголюбов, Медицинская реабилитация (руководство, в 3 томах). // Москва — Пермь. — 1998.
- Попов С. Н. Физическая реабилитация. 2005. — С.608.
Наши специалисты

Врач-невролог, кандидат медицинских наук

врач-невролог, кандидат медицинских наук

Заведующий отделением восстановительной медицины, врач по лечебной физкультуре, врач-невролог, врач-рефлексотерапевт

Врач-физиотерапевт, кандидат медицинских наук

Инструктор-методист по лечебной физкультуре, кинезитерапевт

Инструктор-методист по лечебной физкультуре
Цены на услуги *
*Информация на сайте носит исключительно ознакомительный характер. Все материалы и цены, размещенные на сайте, не являются публичной офертой, определяемой положениями ст. 437 ГК РФ. Для получения точной информации обратитесь к сотрудникам клиники или посетите нашу клинику.
Скачать прайс на услуги
Мы работаем круглосуточно
Миотонический синдром. Симптомы и лечение :: SYL.ru
Каждый родитель желает, чтобы его ребенок рос здоровым, но, к сожалению, не всегда удается избежать каких-либо отклонений. К редким наследственным патологиям относится миотонический синдром, проявляющийся не только у грудных детей, но и в более старшем возрасте. Самостоятельно диагностировать недуг довольно сложно, поэтому без помощи хорошего педиатра и невропатолога не обойтись.
Что собой представляет патология?
Миотонию некоторые специалисты вообще не относят к заболеваниям, называя такое состояние симптоматическим проявлением нервно-мышечных нарушений. Распознать отклонение в развитии, особенно в первый год жизни малыша, практически невозможно. Главная особенность – длительный спазм мышц, который наблюдается после очередного движения или напряжения. Родителям необходимо внимательно следить за тем, как развивается ребенок, и регулярно посещать детского врача. Также важно осознать, что для того, чтобы малыш мог развиваться наравне с остальными детьми и жить полноценной жизнью, понадобится приложить максимум усилий и постоянно заниматься данной проблемой.
Всему виной гены!
В настоящее время установить истинную причину миотонического синдрома ученым не удалось. Врожденный недуг (классическая форма) передается по аутосомно-доминантному типу, хондродистрофическая миотония – по аутосомно-рецессивному. Это значит, что в первом случае измененный ген начинает доминировать и подавлять нормальную копию, тем самым вызывая развитие генетических отклонений. Вероятность того, что ребенок родится с врожденным недугом (у больного родителя) – 50%. Не стоит отчаиваться, ведь каждый малыш имеет шанс появиться на свет абсолютно здоровым, если ему передастся здоровая копия гена. Врожденный миотонический синдром у детей может проявляться в разной степени, но встречается он в каждом поколении.

По аутосомно-рецессивному типу наследуется синдром Шварца-Джампела (хондродистрофическая миотония). Для того чтобы недуг себя проявил, необходимо получить от каждого из родителей по одной измененной копии гена. Если же ребенок унаследует только одну «неправильную» копию, а вторую - здоровую, он будет являться только носителем.
Другие причины
Неврологический миотонический синдром в большинстве случаев является наследственным недугом, но вызвать его могут и не «генные» причины, среди которых:
- Родовые травмы.
- Перинатальная энцефалопатия.
- Нарушение обмена веществ.
- Малая двигательная активность в детском возрасте.
- Перенесенный в младенчестве рахит.
- Нервно-мышечные заболевания.
- Гиподинамия (снижение активности) в период беременности.
Синдром, который развился вследствие возникновения этих причин, считается приобретенным и хорошо поддается лечению, в отличие от генетического типа недуга.
Виды миотонического синдрома
Медицине известны следующие разновидности миотонии:
- Миотония Томсона (Томсена) – аутосомно-доминантный тип недуга, который передается от одного родителя. Впервые заболевание было описано детским врачом Асмусом Юлиусом Томсеном, обнаружившим его у себя и членов своей семьи. Первые симптомы отклонения могут проявиться еще в грудном возрасте, но бывают случаи, когда недуг дает о себе знать только в 6-12 лет.
- Миотония Беккера – передается по аутосомно-рецессивному типу и встречается намного чаще предыдущего вида. Развитие признаков недуга у девочек происходит раньше (4-12 лет), чем у мальчиков (16-18 лет).
- Синдром Шварца-Джампела – проявляется уже в первый год жизни. Ребенок отличается врожденными аномалиями скелета, черепа, умственной отсталостью. Синдром передается на генетическом уровне только от обоих родителей.
- Дистрофическая миотония – ее еще называют болезнью Куршмана-Штейнерта. Объединяет в себе отклонения нервно-мышечного характера и патологии глаз, вегетативной и эндокринной системы.
- Парамиотония врожденная – встречается редко и передается по аутосомно-доминантному типу. Сочетает в себе симптомы периодического паралича и миотонии.
Миотонический синдром: симптомы
Основной признак отклонения, который объединяет все разновидности недуга – спазм мышц. Заключается он в том, что после резкого сокращения мышца не может расслабиться сразу, а только после нескольких повторений движения. Тоническое напряжение остро проявляется после недлительного отдыха. Первые шаги даются больному с трудом, так как для того, чтобы мышца расслабилась, необходимо не менее 30 секунд.

Для выявления нарушений больного просят сжать, а затем быстро разжать кулак или пройтись по лестнице. С первых попыток это сделать не удается, но с каждым разом тонус будет уменьшаться до тех пор, пока мышца полностью не расслабится. В некоторых случая, например, при миотонии Томсена, создается видимость атлетического телосложения и хорошо развитой мускулатуры у пациента. Это вызвано гипертрофией некоторых мышечных групп.
При парамиотонии симптоматика проявляется при воздействии холода и полностью исчезает в тепле. Спазм может длиться от нескольких минут до 2-3 часов, слабость мышц (после применения тепла) наблюдается в течение нескольких суток. При диагностике с помощью электрического тока характерных миотонических явлений не наблюдается.
Миотоническая дистрофия характеризуется проявлением атрофии мышц шеи и лица, а характерные для синдрома спазмы наблюдаются в жевательных мышцах, дистальных отделах конечностей. Мужчины часто страдают облысением, умственной отсталостью.
Рефлекторный миотонический синдром (мышечный спазм) чаще всего возникает в грушевидной и передней лестничной мышце (расположена на шее).
Как выявить отклонение у детей?
Распознать отклонения могут родители у новорожденного малыша. Миотонический синдром у грудничка проявляется даже при сосательных движениях. Такие детки начинают намного позже держать голову, переворачиваться со спины на живот, ползать и ходить. Мышечная слабость приводит к развитию неправильной осанки, сколиозу, возникновению проблем с желудочно-кишечным трактом (запоры, колики).

Миотонический синдром у детей проявляется и тем, что не позволяет вовремя делать первые шаги. Малыши часто падают, теряют равновесие, не могут быстро бегать и ходить по лестнице из-за мышечного спазма. Играть в подвижные активные игры им также не под силу – дети быстро устают.
Слабость лицевых мышц влияет на способность нормально разговаривать. Уже в раннем возрасте проявляются признаки близорукости, так как синдром поражает и глазные мышцы. Симптоматика недуга у каждого ребенка проявляется в разной степени. Следует учитывать, что первые проявления могут возникнуть только в подростковом возрасте. Определить синдром можно попытаться самостоятельно. Для этого по икроножной мышце необходимо легонько постучать, и если произойдет спазм (иногда даже болезненный), значит, понадобиться консультация врача-невролога.
Диагностика
Дать заключение о том, что ребенок страдает миотонией, может только специалист. Для более полной картины врачу необходимо знать о наличии аналогичного синдрома у ближайших родственников. Наиболее простым способом обнаружения дефекта является простукивание перкуссионным молоточком. В качестве инструментального метода диагностики используется электромиография. Метод позволяет врачу установить степень поражения скелетных мышц путем регистрирования их электрической активности.
Парамиотония диагностируется с помощью холодовой пробы, которая вначале вызывает появление миотонических разрядов, после чего любые феномены, характерные для синдрома, исчезают.
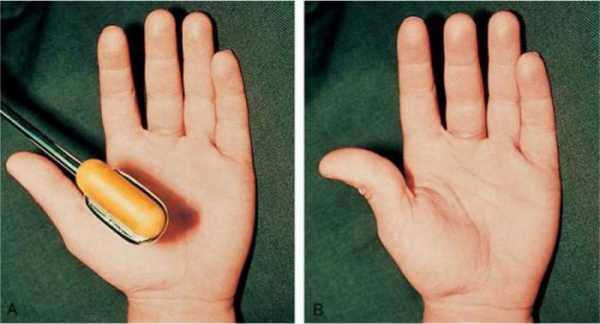
Заключительным этапом диагностики является анализ крови на наличие генетических патологий. Его проводят новорожденным детям при наличии различных жалоб родителей (плохой набор веса, отставание в физическом развитии, частые срыгивания) и более взрослым пациентам.
Проявление недуга у взрослых
В юношеском и зрелом возрасте могут проявиться признаки дистрофического типа миотонического синдрома (болезнь Штейнера). Довольно часто это сопровождается сопутствующими патологиями сердца, нарушением менструального цикла у женской половины пациентов и импотенцией у мужчин. Помимо неврологических отклонений, наблюдается значительное снижение уровня интеллекта.
Миотонический синдром у взрослых лиц проявляется весьма своеобразно. Мужчины (более подвержены синдрому) с гипертрофией мышц выглядят спортивно и подтянуто, но на самом деле такие мышцы не обладают силой. Миотонические спазмы приводят к изменению голоса, трудностям при глотании и дыхании (поражение дыхательных мышц). Первые спазмы мышц обычно поражают ноги. При этом больной может абсолютно нормально ходить по ровной поверхности, но если ему понадобится подняться по ступенькам, то есть задействовать другую группу мышц, преодолеть первый подъем будет довольно сложно.
У пациента с миотоническим синдромом можно наблюдать феномен приседания. Ему не удастся присесть так, чтобы задняя поверхность бедер соприкасалась с голенью и при этом полностью опираться на стопу. Больной не в состоянии удержать равновесие в таком положении и просто упадет. Обычное для здорового человека полное приседание он выполняет с широко расставленными ногами и опирается на носки (полностью присесть при этом все равно не получается).
Миотонический синдром: лечение
Чем раньше будет обнаружено отклонение и начато лечение, тем больше шансов у пациента на нормальное развитие. Обнаружив симптомы нарушения работы мышц у ребенка, необходимо незамедлительно обратиться к педиатру или неврологу. В процессе диагностики специалисты прежде всего должны выявить этиологию недуга, а затем подбирать подходящие меры лечебного воздействия.
Для того чтобы малыш не отставал в физическом развитии, понадобится пройти курс (не один!) физиопроцедур и лечебного массажа. При положительных результатах в дальнейшем ребенок будет посещать специальные занятия ЛФК. Даже при врожденном синдроме эти меры значительно облегчают состояние. Родителям придется научиться делать массаж для того, чтобы в домашних условиях поддерживать здоровье ребенка.
Для улучшения самочувствия понадобиться придерживаться диетического питания (ограничивается потребление солей калия). Важно избегать переохлаждения, стрессовых ситуаций, длительного нахождения в расслабленном состоянии.
Медикаментозное воздействие
Некоторые специалисты придерживаются мнения, что лечить миотонический синдром лекарственными препаратами абсолютно не эффективно, и положительный результат возможет только при постоянных физических занятиях. Определять нагрузку должен только специалист. Но для того чтобы снять спазм мышц в некоторых случаях назначают препарат «Фенитоин». Это мышечный релаксант, обладающий противосудорожным действием. Суточная доза средства рассчитывается в зависимости от веса и возраста пациента. У взрослых она не может превышать 500 мг, у детей – 300 мг в сутки.

При миотоническом синдроме также могут быть назначены следующие препараты:
- «Актовегин» – способствует повышению энергетического ресурса клеток, улучшает процессы метаболизма. Препарат можно применять как для внутривенного введения (инъекции), так и в форме таблеток.
- «Пантогам» – эффективное ноотропное средство, активизирующее метаболизм в клетках и имеющее противосудорожное действие. Справляется с психомоторной возбудимостью, неврозами. Хорошо зарекомендовал себя в лечении умственной отсталости у детей.
- «Элькар» – капли, повышающие мозговую активность и нормализующие процессы метаболизма. В списке показаний к назначению присутствует хроническая мышечная слабость, наследственные патологии мышечной дистрофии.
В обязательном порядке больным с миотоническим синдромом назначают диуретики. Они снижают уровень калия в плазме крови, который обычно повышается после очередного приступа.
Рекомендации родителям
Врожденные болезни нервной системы требуют постоянного поддерживающего лечения. Для того чтобы уменьшить симптоматику проявления недуга, врачи рекомендуют заниматься плаванием. Маленькому ребенку достаточно пространства будет и в домашней ванне, а в старшем возрасте можно посещать бассейн.
В поликлинике в обязательном порядке ребенку назначают физиотерапевтические процедуры. Отказываться от них не желательно, потому как электрофорез действительно приносит облегчение деткам, страдающим от миотонии. Метод абсолютно безболезненный и предполагает местное введение лекарств посредством постоянного тока.

Необходима консультация врача-генетика и при планировании беременности. В зоне риска находятся мужчины и женщины, в чьем роду уже встречались хромосомные отклонения. Настоятельно рекомендуется таким парам проходить генетическую диагностику во время самой беременности. Для этого применяется анализ околоплодных вод (амниоцентез) на 14-18 неделе, биопсия хориона на 10-12 неделе, кордоцентез (проводится после 18 недель). Стоит отметить, что риск рождения ребенка с таким отклонение значительно повышается, если женщине более 35 лет. С помощью генетического исследования крови врачи могут определить предрасположенность будущего малыша к серьезным недугам.
Профилактика
Еще в период беременности женщина, находящаяся в зоне риска, обязана наблюдаться у врача и проходить обследования, помогающие обнаружить генетические патологии. Новорожденному малышу необходимо чаще находиться на свежем воздухе, делать массаж (профилактический). Важно принять меры для предупреждения развития рахита.
Миотонический синдром - Спазмы(судороги)
Виды миотонического синдрома
 Миотоническим синдромом называется феномен расслабления мышц после их сильного напряжения. Существенные усилия при высоком уровне двигательной активности провоцируют дальнейшее развития указанного синдрома. Расслабляющая фаза после того, как произведено чувствительное усилие растягивается по времени на полминуты. Миотонический синдром предполагает существенные затруднения двигательной активности больного после совершения первых же движений, в дальнейшем ему становится легче. Движения его приобретают дополнительную лёгкость и по прошествии определённого промежутка времени происходит их нормализация.
Миотоническим синдромом называется феномен расслабления мышц после их сильного напряжения. Существенные усилия при высоком уровне двигательной активности провоцируют дальнейшее развития указанного синдрома. Расслабляющая фаза после того, как произведено чувствительное усилие растягивается по времени на полминуты. Миотонический синдром предполагает существенные затруднения двигательной активности больного после совершения первых же движений, в дальнейшем ему становится легче. Движения его приобретают дополнительную лёгкость и по прошествии определённого промежутка времени происходит их нормализация.
Можно выделить следующие основные виды миотонического синдрома:
- конгенитивная миотония. Одна из самых распространённых форм синдрома. Заболевание вызывается генной мутацией, обусловленной изменениями структуры гена, несущего ответственность за ионные каналы волокон мышц. Вид синдрома не оказывает существенного влияния на продолжительность жизни и структуру костей;
- конгенитивная парамиотония. Продолжительные сокращения мышц обуславливают такое нарушение. Заболевание сохраняет интенсивность своего развития и течения на протяжении всей жизни человека. Обострение заболевания наблюдается в период повышения нагрузок на организм в холодное время года;
- синдром Шварц-Джемпела. Вид синдрома, наблюдающийся сравнительно нечасто. Имеет особенность передаваться по наследству, проявляясь уже в первый год жизни человека. Характеризуется нарастанием мышечного тонуса.
Миотонический синдром причины
Миотонический синдром может быть врождённым заболеванием и проявиться уже в первые месяцы жизни человека. К основным причинам его возникновения врачи относят поражения ядер подкорки головного мозга, патологию мозжечка и сбои в работе вегетативных нервных центров. Если же проявления синдрома на обусловлены генными причинами, можно сделать вывод что его появление обусловлено детскими патологиями, такими, как рахит, сбои течения процесса обмена веществ, травмы, полученные при родах, нарушения внутриутробного развития плода. Необходимо отметить, что встречаются случаи, когда первоначальный диагноз не подтверждается, при этом может иметь место неправильная интерпретация тонуса мышц.
Миотонический синдром симптомы
Основным симптомом болезненного состояния выступает спазм мышц. После того, как мышца претерпела резкое сокращение её не получается расслабить сразу же, для этого необходимо некоторое количество повторяющихся движений. Непродолжительный отдых также способствует острым проявлениям тонических напряжений. Самые первые движения больной проделывает с величайшими затруднениями, для полного расслабления мышц необходимо не менее полминуты.
Симптомы миотонического синдрома, проявляющиеся на холоде, могут полностью исчезнуть при попадании в тепло. Продолжительность мышечного сокращения варьируется от двух трёх минут до нескольких часов. После этого на протяжении нескольких суток может присутствовать слабость в мышцах, после прекращения воздействия теплоты на организм. Диагностика, проводимая посредством применения направленного на человека воздействия электрического тока, не вызывает определённых проявлений миотонии.
Для миотонической дистрофии характерна атрофия развития мышечных групп области лица, шеи, а также для описываемого синдрома возможны проявления спазмов жевательных мышц, дистальных отделов конечностей.
Рефлексы миотонического синдрома нередко проявляются в передней лестничной мышце, которая имеет локализацию в шейном отделе позвоночника, а также в грушевидной мышце.
Миотонический синдром у ребенка
Миотонический синдром относится к числу болезней мышечной и нервной систем. Его проявления заключаются в понижении мышечного тонуса, что означает сложности с расслаблением мышц различных групп после того, как оно испытали сильное напряжение. Сокращение мышцы можно наблюдать после того, как будет сначала нанесен лёгкий удар по мышце при помощи пластмассового молотка или пальцем. Сокращение может быть весьма длительным и неприятным. В какое либо иное время мышцы ребёнка могут пребывать в пониженном тонусе.
Вялая осанка ребёнка может являться следствием его мышечной слабости, которая сопутствует проявлению миотонического синдрома. Дети с подобными проявлениями начинают двигаться и ходить значительно позже своих сверстников, поскольку им нелегко ровно держать спину и они испытывают боли в суставах, вызванные нарушениями их функционирования. Вследствие нарушения мышечного тонуса и говорить такие дети начинают значительно позже. Их речь при этом невнятна и невыразительна.
Миотонический синдром у взрослых
Для чёткого определения проявления миотонического синдрома у взрослого человека необходимо проведение врачом физиакального обследования посредством разных тестирований, одним из которых можно назвать сжатие и разжатие мячика. Очень важно при этом точно установить, является ли миотонический синдром прогрессирующей болезнью, или же имеют место проявления атрофической миотонии либо какой-нибудь иной болезни. При миотоническом синдроме, в целях облегчения ситуации для больного и назначения правильного курса лечения рекомендуется проведение исследований в лаборатории. Электромиография способна зарегистрировать параметры электрической активности мышц скелета, кроме того требуется проведение биопсии мышц, генетического анализа, исследований крови.
Миотонический синдром лечение
От того, насколько быстро был обнаружен миотонический синдром, зависит скорость, с которой он будет устранён и излечен. Некоторые врачи разделяют мнение относительно неэффективности терапии миотонического синдрома при помощи медицинских препаратов, однако регулярные занятия физкультурой способны оказать надлежащий эффект. Нагрузка при этом должна быть распределена специалистом.
Препараты
В определённых случаях для снятия мышечных спазмов можно применять препарат Фенитоин. Он представляет собой мышечный релаксант, действие которого относится к числу противосудорожных. Расчёт необходимой дозировки производится в соответствии с весом больного человека и его возраста. Для взрослого человека суточная дозировка недолжна превышать 500 мг, для ребёнка 300 мг в течение суток. Кроме того при миотоническом синдроме можно рекомендовать следующие препараты:
- Актовегин. Повышает энергетический потенциал клеток организма человека, ускоряет течение метаболизма. Имеется в виде таблеток, также можно вводить внутривенно;
- Пантогам. Также вызывает активизацию процесса метаболизма в клетках организма, действие его противосудорожное. Устраняет возбудимость и неврозы. С его помощью проводят профилактику и лечение задержек умственного развития у детей;
- Элькар. Представлен в виде капель, оказывающих воздействие на процессы активности мозга и ускоряющих метаболизм. Рекомендован также при слабости мышц и дистрофии.
Массаж
Массаж при описываемом синдроме эффективен, если он проводится достаточно интенсивно. Сперва необходимо тщательно размять массируемую область, после чего тщательно и интенсивно помассировать на протяжении 15-20 минут. В конечном счёте решающее значение имеет интенсивность проведенных операций. Рекомендуется проводить массаж ежедневно на протяжении недели для существенного облегчения состояния больного.
Электрофорез
Проведение электрофореза относится к разряду процедур физиотерапии. Его суть заключается в том, что применяемое при лечении вещество аккуратно наносится на электродные прокладки и посредством воздействия магнитного поля вводится в организм человека. Именно в месте введения средства оказывается наибольшее воздействие на процессы, приводящие к миотоническому синдрому. Электрический разряд известен своим гуморальным и нервно-рефлекторным воздействием.
Гимнастика
Выполнение гимнастических упражнений обеспечивает должный уровень двигательной активности всех мышечных групп организма. Соответствующий подбор необходимых упражнений рекомендуется проводить, консультируясь с грамотным специалистом в этой области, однако следует заметить, что интенсивность упражнений следует увеличивать постепенно, во избежание перегрузки подвергшихся заболеванию участков.
Похожие статьи:
Боли в животе спазмами
Таблетки снимающие спазм
Блефароспазм
Как снять спазм мышц
Спазм при различных состояниях
Миотонический синдром. Клиника Бобыря
27 авг. 2019Миотонию трудно назвать болезнью в классическом понимании. Это симптом нарушений деятельности мышечной системы, выражающийся в сокращениях мышц, вызванных повышенным их тонусом, и в невозможности быстрой мышечной релаксации. Приступ миотонии, или миотонич
Показателями приступа являются сложности, которые возникают при попытке встать, а также чувство напряженности. Повышенный тонус влечет за собой медленную релаксацию. Миотонический синдром в широком понимании - это любая нарушенная релаксация усиленного тонуса в мышцах, которое не относится ни к пирамидному, ни к экстрапирамидному гипертонусу.
Длительность миотонического приступа составляет от нескольких десятков секунд до нескольких десятков минут. Интенсивность приступов варьируется от легкой скованности до частичной блокировки двигательной функции. Поводом для приступов могут послужить как сильные физические нагрузки, так и долгое пребывание в состоянии расслабленности. Также реакцию могут вызвать холод или сильные шумы. Заболевание поражает только скелетные мышцы. Соответственно, приступ - это только результат осознанных движений, т.е. мышечные волокна, участвующие в работе органов, никак не связаны с этой болезнью. Миотония проявляется в сокращениях мышц ног, лица, рук, плечевого пояса. В редких случаях данная болезнь может отразиться и на способности говорить.
Миотоническим синдромом может заболеть любой человек, поскольку обусловлен генетической детерминированностью. Первые приступы могут появиться в любое время, вне зависимости от возраста. Первые симптомы заболевания, согласно данным исследований, проявляются уже в ранней юности, однако болезнь может иметь и отложенное действие и проявиться, к примеру, уже в более зрелом периоде. Наследование происходит по аутосомально-доминантному или аутосомально-рецессивному пути.
Аутосомально-доминантное наследование
Передающийся таким путем дефект всегда затрагивает все новые поколения. При «аутосомальном» типе дефектный ген может находиться в любой клетке, кроме половых. В данном варианте одного носителя вполне хватит, при этом не имеет значения, мама это будет или отец. В данном случае каждый ребенок от любого родителя-носителя может родиться с генетической ошибкой с вероятностью 50%. Возраст проявления и развития заболевания носят в данном случае индивидуальный характер.
Аутосомально-рецессивное наследование
Рецессивное наследование предполагает проявление генетического дефекта у одного поколения без обязательной «семейственности» нарушения. Для проявления генетического дефекта необходимо, чтобы двое родителей имели дефектный ген. При этом у них болезнь может так и не проявиться. В 25% случаев есть вероятность наследовать генетическую ошибку от двух родителей. В случае, когда носителем является только один родитель, то нарушение может также быть унаследовано ребенком. И он в свою очередь также станет носителем дефектного гена, однако признаки, вероятно, не проявятся. При таком положении есть вероятность 50% того, что новорожденный унаследует дефект. Однако существует 25% того, что наследования не произойдет.
Виды миотонии. Миотония конгенита
Этот вид заболевания один из самых распространенных регрессивных форм синдрома. Причина заболевания связана с мутацией гена, который отвечает за натриевые ионные каналы мышечных волокон. Болезнь не влияет на продолжительность жизни, а также практически не меняет внешность или форму скелета. Данный вид миотонии подразделяется на два подвида по типам наследственности.
Выраженная генерализованная миотония Беккера, передающаяся по аутосомально-рецессивному типу, встречается чаще остальных видов, является довольно тяжелой формой болезни. Данный тип носит имя немецкого генетика Петера Эмиля Беккера, который впервые дал описание болезни. Заболевание развивается от 4 до 12 лет у девочек и около 18 лет у мальчиков. Интенсивность проявления симптомов может усиливаться агрессивно несколько лет после утверждения диагноза или же более медленно вплоть до достижения больного возраста 20 лет.
Наследственное заболевание Томсена, передающееся аутосомально-доминантному типу, сопровождается длительными тоническими спазмами мышц. Болезнь носит имя датского врача Асмуса Юлиуса Томсена. Сам доктор обнаружил у себя и своей семьи это заболевание и стал исследовать его течение на самом себе. Как правило, впервые болезнь дает о себе знать в возрасте 6-10 лет и выражается в длительных неболезненных мышечных сокращениях длительностью до нескольких десятков секунд. Симптомы данной болезни обычно не такие тяжелые, как при заболевании Беккера. Бывают, однако, случаи, когда симптомы едва ли проявляются на протяжении нескольких лет после обнаружения заболевания.
Для диагностирования можно обратиться к очень простому тесту – спуск и подъем по лестнице. Трудности во время перемещении по ровной поверхности могут возникнуть, если до этого больной находился в длительном покое, а также вследствие изменения темпа движения. Спазмы в мышцах рук могут стать причиной проблем в процессе письма или при рукопожатии. Также могут возникать сложности в произнесении первых нескольких слов, в процессе глотания, при первых жевательных движениях и прочее. Спазмы могут возникать и в круговых мышцах глаз.
И болезнь Томсена, и болезнь Беккера характеризуются генерализованной миотонией, вызванной произвольными сокращениями. Связано это в основном с повышенной физической нагрузкой или же, наоборот, с длительным отсутствием движения и расслабленности в мышцах. Болезнь особенно выражена в ногах, что проявляется в затруднениях во время ходьбы, а порой может вызывать падения. Заболевание также выражается в скованности плечевого мышечного пояса, мышц головы. У больных могут возникать проблемы с процессами жевания, мигания или хватания предметов. Также последствиями приступов может быть обездвиживающая слабость.
После приступов в обоих случаях миотонии снизить напряженность в пораженных областях проще всего путем повторения движений. Приступы проявляются в интенсивных сокращениях мышц. После приступа нужно несколько раз активизировать сокращенные мышцы и после пяти повторений уровень напряженности начнет спадать. Это называется эффектом разминки, что позволяет больным заниматься определенными силовыми видами спорта.
В обоих случаях у больных наблюдаются нестандартные размеры мышечного пояса, в особенности на ногах, ягодицах, руках, плечах и спине. Порой больные этими заболеваниями похожи на реальных культуристов, однако, на самом деле это результат гипертрофии мышц. Последнее особенно заметно при миотонии Беккера.
Парамиотония конгенита
Это редкое конституциональное нарушение, характеризуется длительными мышечными сокращениями. Болезнь вызвана нарушениями натриевых каналов, передается по аутосомно-доминантному типу. Интенсивность заболевания сохраняется на протяжении всей жизни. Заболевание проявляется на ранних этапах жизни. Тонические сокращения мышц охватывают руки, лицо, шею. Толчком, вызывающим болезнь, может послужить повышенная степень нагрузок, а также холод. В последнем варианте приступ можно унять, согревая подвергнутое приступу место.
Как правило, во время приступов мышечных сокращений наблюдается обездвиживание и повышенная слабость соответствующих областей. При этом слабость может продлиться и после приступа, до нескольких часов.
Однако при парамиотонии конгенита слабость отсутствует. Также для парамиотонии свойственно увеличение мышечной напряженности, возникающей после приступа, это значительно уменьшает способность двигаться. В медицинской практике такое явление носит название парадоксальной миотонией.
Синдром Шварц-Джемпела
Этот вид еще называют миотонической хондродистрофией. Встречается довольно редко, передается по наследству по аутосомально-рецессивному типу. Первые симптомы заметны обычно уже в первый год жизни, сопровождается нарастающим и усиливающимся тонусом мышц. Спазмы вызваны спонтанными высокочастотными разрядами мышечных волокон и периферийных нервов. При данном заболевании возникают мышечные спазмы по большей части в лице и бедрах. Также наблюдается нарушение речи с изменениями в конфигурации лица. Люди с синдромом Шварц-Джемпела страдают разными аномалиями скелета, которые, как правило, приводят к нарушениям процесса роста. Помимо этого, внешне болезнь проявляется низкопосаженными ушами, высоким нёбом, блефарофимозом, в маскообразном лице с большим количеством морщин в области рта, в подбородке, скошенном от напряжения. Бывают случаи, когда у пациентов проявляется снижение мозговой активности.
По какой причине возникает синдром Шварца-Джемпела, неизвестно. Предположительно, это один из видов нарушений мышечной деятельности. Также синдром объясняют возможным сочетанием нарушений нервно-мышечной возбудимости.
Диагностика
Диагноз миотонии в принципе врач сможет установить и при физиакальном обследовании путем различных тестов, в том числе такой стимуляцией, как сжимать и разжимать мячик. Однако крайне важно определить, является ли это прогрессирующим заболеванием, таким как атрофическая миотония или синдром Шварц-Джемпела, или же это миотонический синдром. В этом случае необходимо пройти лабораторные исследования, например, электромиография. С помощью ЭМГ регистрируется электрическая активность скелетных мышц. Также нужны исследования крови, генетический анализ, мышечная биопсия.
Лечение
Сегодня возможность на сто процентов избавиться от любого из перечисленных миотонических синдромов не существует. Интенсивность и способы лечения зависят от симптомов. При повышенной интенсивности и частоте атак назначают специальные лекарственные препараты, блокирующие симптомы. Среди назначаемых препаратов можно назвать такие, как мексилитен, гуанин, прокаинамид, тегретол, фенитоин. Существенным минусом этих лекарств является наличие большого количества побочных действий, и злоупотреблять ими не рекомендуется. Более действенный способ – выявить, что именно может служить толчком к возникновению симптомов и избегать этих моментов. Если все же миотонической атаки избежать не удалось, то лучше помочь восстановлению мышц спокойным отдыхом.
Автор: К.М.Н., академик РАМТН М.А. БобырьМышечно-тонический синдром: причины, симптомы, лечение
Мышечно-тонический синдром — сложный симптомокомплекс, обусловленный перенапряжением мышечных волокон, появлением в их толще болезненных и плотных тяжей. Это компенсаторное повышение тонуса мышц, возникающее в зоне иннервации пораженного сегмента позвоночника. Спазм мышц происходит спонтанно при сдавливании и раздражении определенного нервного волокна. Причиной развития недуга чаще всего являются дегенеративные и дистрофические явления в позвоночнике, протекающие в форме остеохондроза или спондилеза.
Заболевание чаще всего развивается при поражении шейных структур. Его основным проявлением является боль — цервикалгия. Болевой синдром имеет резкий характер. Он усиливается при движении головой и сопровождается цефалгией, головокружением, снижением остроты зрения. Шейный отдел позвоночника поражается намного чаще грудного и поясничного сегментов. Его позвонки высоко подвижны и окружены обилием мышц. Они способны совершать движения в различных направлениях. Сосудисто-нервные стволы шеи отличаются особым расположением, что также играет роль в развитии данной патологии. Депрессии и стрессы провоцируют психовегетативный синдром, который проявляется дистонией мышц шеи, лица и головы, приводящей к появлению боли в голове и прочих симптомов синдрома.
Заболевания органов, расположенных в грудной полости, сопровождаются изменением тонуса и чрезмерным натяжением соответствующих мышц. Патология почек, органов мочевыведения и репродуктивной системы проявляются расстройствами сексуальной сферы, болевым синдромом, перенапряжением мышц крестцово-поясничной области.
Виды патологии:
- Умеренный синдром — боль возникает только при прикосновении, в мышце пальпируются болезненные уплотнения;
- Выраженный синдром — боль становится нестерпимой при каждом прикосновении, ее интенсивность усиливается при растирании пораженной части, мышечные волокна напоминают фиброзные тяжи.
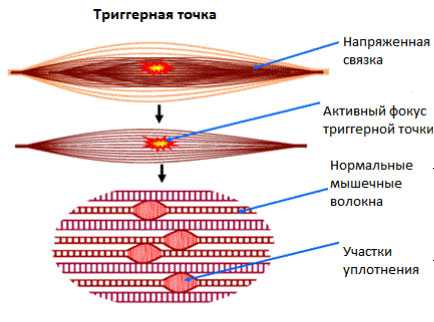
формирование уплотнений в мышцах при прогрессировании мышечно-тонического синдрома
Типы синдрома:
- Локальный — поражение одной мышцы или ее одного участка;
- Диффузный — поражение целой группы мышц.
Этиология и патогенез
Патология имеет вертеброгенное происхождение и является следствием остеохондроза. При раздражении болевых рецепторов возникает дискомфорт, который постепенно перерастает в мучительную и нестерпимую боль. В ответ на ее появление пораженные мышцы спазмируются. Длительный спазм — причина боли. Так замыкается порочный круг, лежащий в основе данного недуга.
Заболевания, проявляющиеся признаками патологии:
- Воспаление позвоночника с ограничением его подвижности,
- Межпозвоночные грыжи,
- Артроз суставов,
- Анкилозирующий спондилит,
- Сколиоз, кифоз, лордоз,
- Миофасциальный синдром,
- Травматическое повреждение позвоночника,
- Воспаление оболочек мозга,
- Геморрагический инсульт,
- Заглоточный или эпидуральный абсцесс,
- Аутоиммунные заболевания соединительной ткани.

Факторы, способствующие развитию патологии:
- продолжительное нахождение в неправильном положении,
- нарушение осанки,
- гиподинамия,
- физическое перенапряжение,
- сквозняки,
- всплески эмоций,
- гиповитаминоз,
- табакокурение,
- деформация нижних конечностей,
- лишний вес,
- несбалансированное питание.
Шейный мышечно-тонический синдром часто развивается у лиц, работающих за компьютером, у офисных работников и всех, кто ведет малоподвижный образ жизни, трудится в одной и той же позе с фиксированным положением головы. Тяжелая верхняя одежда, узкий ворот рубашки и туго затянутый галстук оказывают давление на структуры шеи, что также способствует развитию патологии.
Развитию синдрома поясничного отдела способствует чрезмерно активное перекапывание земли, подъем тяжестей, долгие пешие прогулки. Провоцирует его появление также неудобная постель, атипичная постановка стопы, не по размеру подобранная обувь.
Патогенетические звенья патологии:
- Дегенеративно-дистрофические изменения в позвоночнике,
- Раздражение рецепторов боли, расположенных около межпозвоночного диска и вдоль связочного аппарата позвоночного столба,
- Спазматическое сокращение мышц,
- Чрезмерное мышечное перенапряжение,
- Нарушение оттока крови,
- Отечность тканей,
- Нарастание боли,
- Долгое мышечное спазмирование,
- Сдавливание сосудисто-нервных пучков уплотненными мышцами,
- Кислородное голодание,
- Дистрофия,
- Дисфункция пораженных мышц,
- Отмирание мышечных волокон,
- Замена мышц соединительной тканью,
- Рубцевание — образование жестких тяжей,
- Ограничение подвижности позвоночника,
- Усиление проявлений синдрома.
Мышечный спазм сигнализирует о наличии заболевания спины, которое рано или поздно проявит себя. Остеохондроз без лечения неуклонно прогрессирует.
Клиническая картина
Проявления патологии, возникающие при поражении шейного отдела позвоночника:
- Тяжесть и дискомфорт в шее,
- Цервикалгия,
- Трудности при поворотах и наклонах головы,
- Резкая или давящая, тупая головная боль, не поддающаяся действию анестетиков и распространяющаяся от затылочной области к височной,
- Образование уплотнений в мышечной ткани,
- При пальпации мышцы напоминают резиновый шланг,
- Отечность шеи,
- Скачки кровяного давления,
- Хруст в шее, возникающий при движении головой,
- Снижение остроты зрения,
- Расстройство слуха,
- Парестезии рук и лица.

Боль является основным симптомом патологии. Она имеет ноющий, тупой, мучительный характер и большую распространенность. Пациенты могут указать конкретное место локализации боли. При нажатии на болевую точку неприятные ощущения усиливаются. Болезненные чувства переносятся крайне тяжело. У пациентов возникает бессонница из-за постоянного поиска выгодной позы. Они устают, изматываются, впадают в отчаяние, погружаются в депрессию. В тех местах, где боль максимально выражена, образуются очаги уплотнения, обусловленные отложением солей кальция.
Цервикалгия при мышечно-тоническом синдроме имеет различную степень выраженности. Иногда состояние пациента ухудшается настолько, что у него мутнеет в глазах, возникает общая слабость и чувство тяжести в затылочной области, которое больной сравнивает с ощущением шлема на голове. Резкая боль в шее отдает в левую или правую часть головы. Пальпаторно по задней поверхности шеи обнаруживаются плотные и болезненные тяжи. Это довольно плотные и грубые образования, напоминающие на ощупь веретена. По мере прогрессирования патологии больные начинают жаловаться на появление дискомфортных ощущений в плече. Они отмечают локальную боль, ограничение подвижности сустава, повышенную чувствительность кожи плечевой области. Боль в течение дня постоянная, ее пик приходится на утреннее или ночное время. При надавливании пальцем на межпозвоночные связки боль обостряется. Спазмированные мышечные волокна непроизвольно подергиваются. При активном выполнении различных действий плечом появляется характерный хруст. Такой же хруст возникает при ощупывании тяжа на шее. Шея у больных симметричная, но опухшая. В дальнейшем они отмечают онемение, дискомфорт и боль в запястье и пальцах.
При развитии патологии на уровне груди основным симптомом также является боль. Она возникает внезапно и затрудняет полноценный вдох и выдох. Болевой синдром напоминает кардиалгию при заболеваниях сердечно-сосудистой системы. В результате в крови накапливается углекислота, и возникает гипоксия. Симптомы подобных процессов: головокружение, слабость мышц, судороги, апатия.
Гипертонус мышц поясничного отдела позвоночника проявляется мучительной болью, ограничивающей выполнение повседневных действий, а также парестезиями и слабостью в ногах. Больным очень сложно сгибаться и разгибаться, вставать с кровати. При пальпации позвонков боль усиливается.
Диагностические мероприятия
Диагностика патологии заключается в выслушивании жалоб больного и тщательном сборе анамнеза. Специалисты пытаются понять, как долго длится болевой синдром, насколько он интенсивен, какой характер имеет и как взаимосвязаны спазмы и движения?

Определение неврологического статуса пациента — следующий этап в постановке диагноза. Обычно у больных с мышечно-тоническим синдромом отсутствуют общемозговые и очаговые симптомы, менингеальные знаки. Определяется болезненная пальпация краниовертебрального перехода и паравертебральных точек на всех уровнях. Простукивание мышц перкуссионным молоточком позволяет обнаружить дефект.
Инструментальная и лабораторная диагностика:
- Рентгенологическое исследование позвоночника выявляет дегенеративные изменения в костной ткани.
- С помощью томографического исследования можно обнаружить патологические изменения в мышцах и связках.
- Электромиография позволяет выявить имеющееся нарушение нервно-мышечной проводимости.
- Анализ крови на параклинические и биохимические показатели — обязательные стандартные методики.
- Биопсия и гистологическое исследование пораженных мышечных волокон проводится по строгим врачебным показаниям.
Лечение
Диагностикой и лечением патологии занимаются специалисты в области неврологии. Врач после обследования больного ставит окончательный диагноз и назначает лечение по современным медицинским методикам.
Мышечно-тонический синдром неизлечим. Общетерапевтические мероприятия должны быть направлены на основное заболевание, ставшее первопричиной мышечных спазмов. Больным показана симптоматическая терапия, позволяющая вернуть их к нормальной жизни.
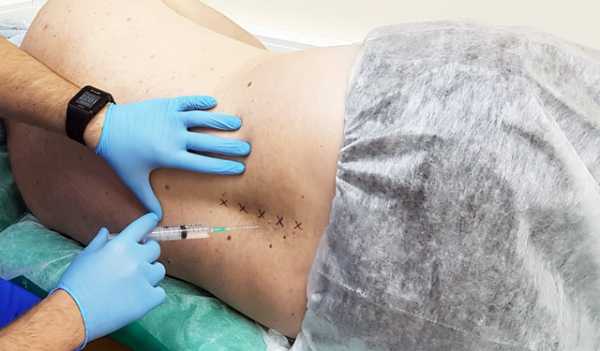
Специалисты назначают пациентам следующие группы препаратов:
- НПВС – «Кетопрофен», «Ибупрофен», «Мелоксикам», «Мовалис»;
- Новокаиновые блокады — введение «Новокаина» в пораженную область;
- Кортикостероиды назначают в виде местных инъекций для снятия боли и прочих признаков воспаления – «Кеналог», «Дипроспан»;
- Миорелаксанты для расслабления пораженных мышц – «Мидокалм», «Сирдалуд»;
- Хондропротекторы – «Алфлутоп», «Терафлекс»;
- Витамины группы В нормализуют обменные процессы в нервной ткани;
- Местно — мази и гели с НПВС: «Вольтарен», «Нурофен», «Долгит»;
- Средства, улучшающие кровоснабжение – «Актовегин», «Трентал»;
- Спазмолитики – «Папаверин», «Дротаверин»;
- Антидепрессанты и антиконвульсанты по индивидуальному врачебному назначению.
Эти лекарства лишь устраняют симптомы. Они не способны полностью восстановить хрящи и оказать благоприятное воздействие на структуры позвоночника. После стихания боли и нормализации мышечного тонуса больные возвращаются к полноценной жизни.
Помимо медикаментозного воздействия широко применяют физиотерапевтические процедуры.
- Массаж устраняет боль, восстанавливает нервно-мышечную проходимость в зоне поражения.
- Специальные корсеты снимают компрессию и разгружают пораженный отдел позвоночника. Они позволяют добиться ортопедического эффекта.
- Всем больным, особенно страдающим цервикалгией, показан сон на ортопедических подушках. С помощью них позвоночник расслабляется и максимально распрямляется.
- Занятия ЛФК и кинезитерапия помогут избежать повторного возникновения синдрома.
- Иглорефлексотерапия нормализует функции нервных волокон и устраняет боль.
- Магнитотерапия, УВЧ-терапия, диадинамические токи и электрофорез усиливают эффект медикаментозного воздействия, уменьшают боль, улучшают кровообращение, нормализуют мышечный тонус.
Если подобное комплексное лечение не дает ожидаемого эффекта, пациенту требуется помощь мануального терапевта или хирурга.
Только устранив источник болезни, можно избавиться от мышечно-тонического синдрома. Почувствовав временное облегчение от приема анестетиков, многие пациенты прерывают курс лечения, а болезнь продолжает прогрессировать. Избавиться от таких патологий, как остеохондроз и другие дегенеративные процессы, очень сложно, а порой и просто невозможно. Именно поэтому следует приложить максимум усилий, чтобы сохранить свое здоровье на оптимальном уровне.
Профилактические мероприятия при данной патологии — занятия посильными видами спорта, ходьба на большие дистанции, правильное питание. Общая физическая подготовка – фундамент здорового тела. Укрепление мышц спины, регулярные физические нагрузки, удобство рабочего и спального места помогают избежать развития синдрома.
Видео: о мышечно-тоническом синдроме при остеохондрозе
Миотония — Википедия
Материал из Википедии — свободной энциклопедии
Миотони́я (от греч. μυος — мышца и лат. tonus — напряжение) или болезнь Томсена[1] — редкое наследственное нервно-мышечное заболевание, характеризующееся длительными тоническими спазмами мышц, возникающими вслед за начальными произвольными движениями[2].
Относится к числу болезней, симптомы которых не исследованы до конца. Типичный симптом — невозможность расслабить в течение нескольких десятков секунд произвольно сокращённые мышцы. Но в ряде наблюдений в случаях несомненной миотонии никаких изменений в мышцах при микроскопическом исследовании не было найдено.
Проявления впервые обнаруживаются в позднем детстве. Путём повторных произвольных сокращений больной постепенно преодолевает спазм, во время которого мышцы при ощупывании резко уплотнены. Миотонические явления значительно усиливаются на холоде и могут существенно ограничить выполнение целого ряда профессиональных и бытовых движений. Спазмы в мышцах ног препятствуют нормальной ходьбе. Обычно наблюдается гипертрофия мышц, придающая больным атлетический вид.
Процесс обмена веществ у больных миотонией каким-то образом нарушен, были отмечены значительные отклонения от нормы в составе мочи больных, но постоянных, специфических для миотонии изменений не обнаружено, так как эти отклонения не одинаковы в различных случаях.
В настоящее время для лечения применяется блокатор натриевых каналов мексилетин, который значимо уменьшает мышечный гипертонус при данном заболевании[3]. Эффективность мексилетина была подтверждена в контролируемом исследовании[4], одном из немногих в области «орфанных» заболеваний.
Некоторое облегчение приносит дифенин, диакарб, хинин. Существуют клинические наблюдения относительно улучшения и даже почти полного излечения болезни. Прогноз для жизни благоприятный. С возрастом миотонические явления ослабевают.
- Шнайдер Н.А., Никулина С.Ю., Шпрах В.В. Миотония. — М: Т.М. Андреева, 2005. — 256 с. — 1000 экз. — ISBN 5-94982-016-9.
Дистрофическая миотония: клиническая картина и лечение
Классификация
Исследователи выделяют две группы миотонических синдромов:
- дистрофические;
- недистрофические.
В каждой группе существует своя дополнительная классификация патологий в зависимости от особенностей проявлений, времени развития.
Дистрофические миотонии
Болезни данной группы объединяются наследованием по доминантному принципу и характеризуются миотоническим, вегетативно-трофическим и дистрофическим синдромом. Особенностью их является отложенное расслабление после напряжения, растущая слабость мышц и их атрофия.
По времени возникновения говорят о конгенитальной, ювенильной, взрослой и поздней форме. Конгенитальная проявляется у ребенка сразу после рождения. Ювенильная – от года до подросткового периода. Взрослая – от 20 лет до 40. Поздняя развивается по достижении сорокалетнего возраста.
В зависимости от того, какой ген мутировал, выделяют дистрофическую миотонию I, II и III типа. К первому относят патологии, которые представляют собой форму, переходную между миотонией и миопатией. Типичный пример – болезнь Россолимо-Штейнерта-Куршмана. Поражаются мышцы конечностей, органов дыхания, миокарда. Уже в детском возрасте появляются нарушения скелета.
Патологии второго типа проявляются у людей разного возраста – от 7 лет до 60. Наблюдается скованность движений, сопровождаемая болями. По мере развития отмечается слабость мышц конечностей, кистей. Развиваются эндокринные заболевания.
Третьему типу свойственна слабость глубоких мышц конечностей, тела. Атрофия мускульной ткани шеи и плеч ведет к свисанию головы. Нарушается внимание, мышление, память.

Недистрофические миотонии
К этому типу относят патологии, связанные с изменением генов натриевого и хлорного каналов. Основным проявлением является слабость в кистях.
Миотонии натриевого канала включают калийзависимую, врожденную парамиотонию, а также гиперкалиемический периодический паралич с миотонией.
К калийзависимому типу относят патологии, передаваемые по рецессивному и доминантному признаку. Возникает этот тип миотонии у детей от 5 лет, взрослых до 55. Характеризуются спазмами, мышечными болями. Сильнее всего поражаются нижние конечности.
Врожденная парамиотония передается по доминантному признаку. Провоцирующим фактором является холод. Важной особенностью является преходящая слабость мышц, которая может длиться несколько дней. Страдают преимущественно жевательные и мимические мышцы.
Гиперкалиемический периодический паралич с миотонией проявляется до 10 лет, наследуется по доминантному признаку. Приступы слабости возникают после приема продуктов с большим содержанием калия. Возникают в ногах, распространяют на тело и руки. Длятся такие эпизоды до двух часов.
К каналопатиям хлорного канала относят миотонию Томсена и болезнь Беккера. Первой свойственно спазмирование мышц пальцев и жевательной мускулатуры. Проявляется в раннем возрасте, в некоторых случаях состояние стабилизируется. В целом мышечная ткань остается достаточно развитой.
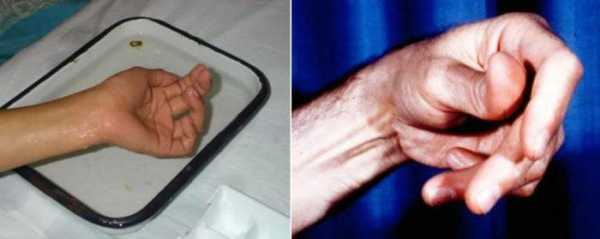
Миотонию Беккера обнаруживают у детей от 4 лет до 18. Имеет более тяжелое течение по сравнению с болезнью Томсена. Характеризуется мышечными болями. Поражаются дистальные, мимические мышцы, мускулатура конечностей.
Этиология
Все виды миотоний обусловлены генетическими нарушениями. В ряде случаев провоцирующим фактором является аутосомно-доминантная передача, в других – аутосомно-рецессивная.
Так, миотония Томпсона наследуется по доминантному принципу, т. е. ген передал ребенку один из его родителей. К этой группе относят также врожденную парамиотонию Эйленбурга, миотонию Россолимо-Штейнерта-Куршмана. По рецессивному типу развивается болезнь Беккера, возникает она из-за передачи мутировавшего гена обоими родителями. Считается, что патологии этого типа проявляются в раннем детстве и имеют более тяжелое течение.
Мутированные гены вызывают нарушение проницаемости мембран клеток, изменения ионных каналов хлора и натрия, медиаторного обмена веществ, что, в конечном счете, ведет к нарушению функций мышечной ткани.
Механизм развития разных видов миотоний един. Ослабленная мускульная ткань из-за воздействия на нее тех или иных факторов приходит в сильный тонус. Возникает состояние, которое называют миотонической атакой. Она появляется в тот момент, когда человек пытается сделать движение, требующее вовлечения пораженных мышечных волокон.
Провоцирующими факторами могут быть стресс, холод, сильные эмоции, длительная обездвиженность.
Считается, что в некоторых ситуациях патологию вызывают кровнородственные связи.
Симптомы
Характерным признаком всех миотоний является симптом «кулака». Характеризуется он тем, что, сжав кулак, больной не может его быстро разжать. Для этого ему потребуется приложить определенные усилия. При последующих сжатиях кулак разжимается легче. Скованность возрастает только при миотонии Эйленберга.
Общие затруднения при всех формах заболевания возникают при попытке открыть рот, встать со стула, быстро открыть глаза, которые до того были закрыты.
Тяжесть проявления симптомов позволяет выделить легкую, среднетяжелую и тяжелую форму заболеваний. Последняя характерна преимущественно для врожденных заболеваний.
Миотония Томпсона и Беккера
В начале развития заболевания возникают болезненные спазмы мышц голени. В дальнейшем поражаются мышцы лица, глотки, языка. Симптомы могут уменьшаться с возрастом. У некоторых они и вовсе исчезают. На смену им приходят парезы и атрофия мышечных волокон головы, шеи. При уменьшении жевательных мышц происходит западение щек. Атрофия шейных волокон ведет к запрокидыванию головы.
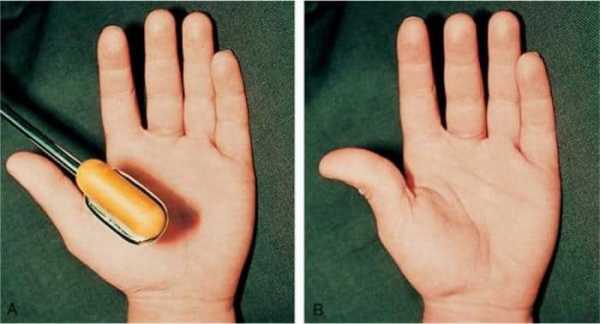
Позже затрагиваются мышцы конечностей. Возрастает их слабость, уменьшается сила.
Страдает сердечно-сосудистая система. Возникают эпизоды аритмии, брадикардии, снижения артериального давления. Выпадают волосы, зубы, кожные покровы становятся очень тонкими.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Первые симптомы проявляются в возрасте 15-20 лет, иногда – в 35. Появляются спазмы мышц, двигательная возбудимость, с развитием болезни эти признаки угасают, чего нельзя сказать о комплексе миопатических симптомов. Развивается атрофия мышечной ткани лица, шеи, кистей рук, снижаются сухожильные рефлексы. Реже поражаются мышцы ног и висков. Постепенно нарастает слабость, больные жалуются на быструю утомляемость.
Атрофия мышц гортани ведет к нарушению глотания, осиплости или потере голоса. У мужчин развивается импотенция, у женщин нарушается менструальный цикл. Часто возникают нарушения сердечно-сосудистой деятельности, ведущие к аритмии, брадикардии.
У многих больных появляется катаракта. Во сне возможны приступы апноэ.
Болезнь Лейдена-Томсена-Беккера
Основной признак – невозможность расслабить мышцы после их напряжения, возникают спазмы. Они поражают человека, когда он закрывает глаза, смыкает челюсти или сжимает руки в кулак. При этом обратное движение в течение долгого времени сделать не получается.
По внешнему виду больные похожи на спортсменов. На ощупь мышцы твердые, плотные, однако силы в них нет.
Миотония хондродистрофической формы
Больных отличает низкий рост, врожденный вывих бедра, скованная мимика, скованность суставов.
Врожденная дистрофическая миотония
Патология характеризуется нарушением у ребенка частоты сердечного ритма, повышенной сонливостью, усилением скованности в холод, эндокринными патологиями.
Парамиотония Эйденбурга
Расслабление мышц затрудняется при холодной температуре окружающего воздуха или местного воздействия. Так, при употреблении сильно холодных продуктов спазм охватывает глотку и язык. Снимается он после согревания.
При общем переохлаждении возникает так называемый «холодный паралич».
Диагностика
Для определения точного диагноза проводится осмотр больного, проверка сухожильных рефлексов, собирается информация о ходе развития патологии, назначаются следующие исследования:
- Электромиография. Регистрируются биоэлектрические импульсы различных участков мышечной ткани, характеризующие поражения нервной системы. Проводится преимущественно электромиографическое исследование.
- ДНК-диагностика.
- Биохимический анализ крови. Обнаруживаются антитела к калиевым каналам, повышенное содержание креатинфосфокиназы.
- Гормональное исследование. Проводится при обнаружении эндокринных нарушениях.
- ЭКГ. Назначается для контроля возникновения и развития сердечно-сосудистых патологий.
Основной целью является дифференциальная диагностика миотонии одного вида от другого.
Лечение
В настоящее время проводится только симптоматическое лечение миотонии. Каких-либо способов полностью остановить течение болезни не существует.
Для снижения судорог и расслабления мышечных тканей назначается Фениотин, Дифенин, Мексилетин. С целью уменьшения содержания калия – диуретики. Подавляют иммунные реакции введением иммуноглобулина. При необходимости применяются анаболитические средства. Тяжелые случаи патологии лечат курсами глюкокортикоидов. Аритмию снимают Новокаинамидом, Хинином.
Некоторым больным назначают курсами препараты, направленные на улучшение обмена веществ (Актовегин), ноотропные лекарства, способные убрать последствия чрезмерного двигательного возбуждения (Пантогам).
Важную роль в предотвращении развития заболевания и облегчении его симптомов играет диета. Она основана на ограничении потребления продуктов, содержащих калий.
Назначается физиотерапия. Основной метод – электромиостимуляция, направленная на стимуляцию нервно-мышечной системы с помощью электрических импульсов.
Рекомендован массаж. Его проводят курсами по 2-3 раза в год.
Несколько раз в год проводится ЛФК с физиотерапевтом. В остальное время показано выполнять физические упражнения в домашних условиях. Рекомендуется плавать в бассейне. Физическая активность помогает нормализовать мышечный тонус, восстановить активность мышц.

Осложнения
Опасность для людей, страдающих миотонией, представляют последствия этой патологии. Среди них выделяется апноэ, пневмония, болезни сердца, аритмия, снижение интеллекта.
Прогноз
Легкие формы патологии не ведут к потере трудоспособности и летальному исходу. В случае развития осложнений, связанных с заболеваниями сердца, возможна смерть от его остановки.
Профилактика
Генетическая обусловленность миотоний не оставляет возможностей для ее профилактики. Единственная возможная мера – проведение ДНК-экспертизы перед планированием беременности. Рекомендована она, прежде всего, тем, чьи родственники страдают от этой патологии.
Миотония представляет собой группу разнородных заболеваний, характеризующихся спазмированием после их напряжения. Патологии носят прогрессирующий характер, однако редко приводят к инвалидизации и гибели. В настоящее время лечение направлено только на снятие остроты симптомов.
Для подготовки статьи использовались следующие источники:
Латышева В. Я., Дривотинов Б. В., Олизарович М. В. // Неврология и нейрохирургия: учеб. пособие — Минск, Выш. шк. 2013.
Коллектив авторов // Нервные болезни — «СпецЛит», 2011 (Учебник для средних медицинских учебных заведений).
Гусев Е. И., Коновалов А. Н., Скворцова В. И. // Неврология и нейрохирургия под ред. Коновалова А. Н., Козлова А. В. — 2014.
причины, симптомы, диагностика и лечение
Нервно-мышечные болезни – это обширная группа генетически гетерогенных наследственных заболеваний нервной системы. Одной из разновидностей такой патологии является миотония. Данное заболевание не однородно, а представлено целым рядом синдромов. В основе патологического процесса лежит изменение работы ионных каналов хлора и натрия. В результате повышается возбудимость мембраны мышечных волокон, клинически проявляемая тонусными нарушениями с постоянной или преходящей мышечной слабостью.

Классификация
Глобально миотонические синдромы делятся на дистрофические и недистрофические. К первой группе относят доминантно наследуемые заболевания, клиническая картина которых складывается из трех ведущих синдромов:
- миотонического;
- дистрофического;
- вегетативно-трофического.
При дистрофических формах наблюдаются отложенное по времени расслабление мышц после их, нарастающая мышечная слабость и дистрофия (атрофия) скелетной мускулатуры. В настоящее время известны три вида дистрофических миотоний, принципиальное отличие которых касается генетического полиморфизма. Ярким примером данной патологии является дистрофическая миотония Россолимо-Штейнерта-Куршмана – наиболее распространенный мышечно-тонический синдром, имеющий наследственный характер.
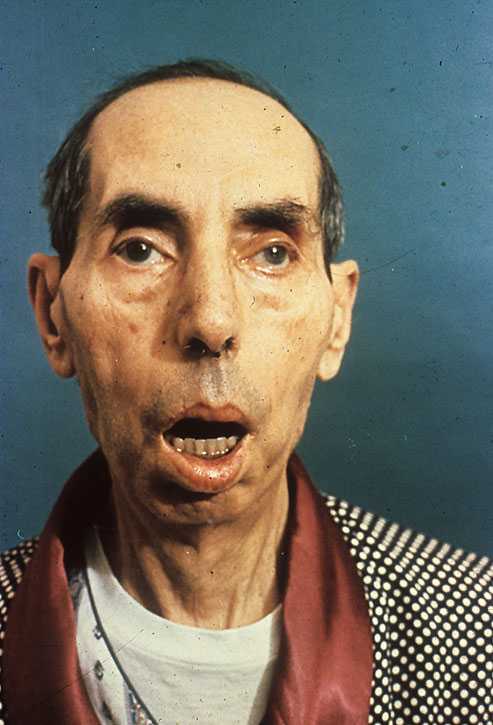
Дебют заболевания дает основание следующей классификации дистрофической миотонии:
- конгенитальная форма – клинические симптомы выражены сразу после рождения;
- ювенильная форма – манифестация болезни от 12 месяцев до подросткового возраста;
- взрослая форма – проявления возникают в возрасте от 20 до 40 лет;
- форма с поздним началом – наиболее легкая форма, проявляется у лиц старше 40 лет.
Недистрофические миотонии градируются в зависимости от мутации генов хлорного или натриевого каналов. К заболеваниям с мутациями гена хлорного канала относятся миотонии Томпсена и Беккера. Патология гена натриевого канала может проявиться в виде различных вариантов калийзависимых миотоний (волнообразной, постоянной, диакарбзависимой), парамиотонии или гиперкалиемического периодического паралича с миотонией.
По некоторым источникам к группе миотоний может быть ошибочно отнесена описанная в 1900 году миатония Оппенгейма, которая характеризуется врожденной мышечной слабостью («синдром вялого ребенка»). Однако данная патология представляет собой непрогрессирующую миопатию с отсутствием миотонических феноменов. Неправильная интерпретация патологии возникает в связи со схожестью терминов «миотония» и «миатония».

Этиология и патогенез
В основе инициации заболевания лежит генетическая поломка, приводящая к патологии ионных каналов хлора и натрия. Миотонии имеют аутосомный тип наследования и могут характеризоваться как доминантной, так и рецессивной передачей мутантного гена. Хромосомный дефект рассматривается как причина извращенного синтеза определенных белковых структур хлорных и натриевых каналов мышечной ткани. Как следствие, нарушается биоэлектрическая активность мембран клеток, провоцирующая чрезмерную возбудимость. При этом периферический мотонейрон может функционировать абсолютно нормально, однако на обычный импульс мышцы реагируют сверхсильным возбуждением, которое блокирует физиологическое расслабление волокон.
Дистрофические миотонии
Дистрофический подтип миотоний – наиболее распространенная группа заболеваний из ряда миотоний. При этом мутация гена затрагивает не только мышечную ткань, но и сердечно-сосудистую систему, орган зрения и головной мозг. Кроме того, характерными проявлениями патологии являются психогенные симптомы – пониженный уровень жизненной мотивации, инертность, тревожно-депрессивные расстройства.
К дистрофической миотонии (ДМ) отнесены три формы болезни, сопровождающиеся, кроме основного клинического синдрома, дистрофическим и вегетативно-трофическим проявлениями, что и является их ключевой особенностью. ДМ представлены тремя типами (формами) в зависимости мутировавшего гена. Так, ДМ 1 типа (болезнь Россолимо-Штейнерта-Куршмана или атрофическая миотония) возникает при мутации 19 хромосомы, 2 типа – 3 хромосомы, 3 типа – 15 хромосомы.
- ДМ 1-го типа относится к аутосомно-доминантным заболеваниям.
Часто ее рассматривают как переходную форму между миотоническими синдромами и миопатиями. Характерным является поражение дистальных отделов конечностей, дыхательной мускулатуры, деформация опорно-двигательного аппарата. Достаточно рано присоединяется двусторонняя катаракта, нарушения сердечного ритма, кардиомиопатия. Миотония Россолимо часто сочетается с патологией желудочно-кишечного тракта и эндокринной системы.
- ДМ 2-го типа дебютирует в возрасте от 7 до 60 лет.
Начало, как правило, постепенное с ограничения и скованности движений, сопровождающихся мышечной болью. В последующем появляется мышечная слабость, более выраженная в проксимальных отделах конечностей и мышцах кисти (преимущественно сгибателях). Дистрофия скелетной мускулатуры умеренная. Поражение мимических мышц не свойственно данному заболеванию. ДМ 2-го типа может сочетаться с развитием сахарного диабета, нарушения толерантности к глюкозе, патологии щитовидной железы, гипогонадизма.
- ДМ 3-го типа развивается с мышечной слабости в мышцах туловища и проксимальных отделах конечностей.
По мере прогрессирования заболевания развивается выраженная мышечная гипотрофия, в патологический процесс вовлекаются дистальные отделы конечностей. Типичен феномен «головы тряпичной куклы», развивающийся за счет атрофии мышц надплечья и шеи. Для данной формы характерны выраженные нарушения высших психических функций – расстройства внимания, памяти, логического мышления.
Недистрофические миотонии
Специфическими симптомами недистрофических миотоний служат изначальная слабость и неловкость в кистях, исчезающая после нескольких повторных произвольных сокращений мышц. Такое клиническое проявление называют «симптом транзиторной слабости», а уменьшение или исчезновение миотонии при повторных мышечных сокращениях – «феномен врабатывания». Миодистрофия не характерна.
Каналопатии с мутациями гена хлорного канала
Каналопатии с мутациями гена хлорного канала имеют синоним – «врожденные миотонии». При аутосомно-доминантном типе наследования данной патологии диагностируется миотония Томпсена. Аутосомно-рецесивную форму недистрофических миотоний называют болезнью Беккера.
- Миотония Томпсена проявляется симптомами активных мышечных спазмов.
Возникают они, главным образом, в жевательной мускулатуре и сгибателях пальцев, появляются при начале выполнения двигательных актов. Достаточно выражен механический мышечно-тонический феномен, когда мышечные сокращения провоцируются внешним воздействием на волокна (например, при ударе неврологическим молоточком.
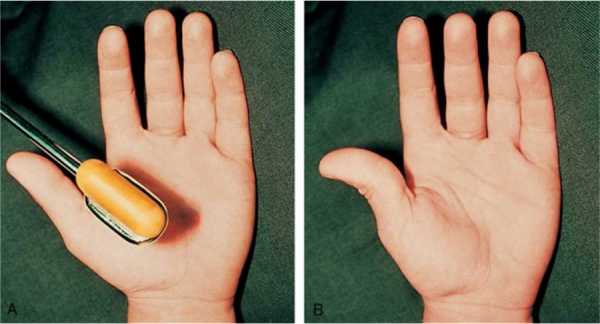
Мышечный корсет при этом развит хорошо, мышцы твердые на ощупь, находятся в постоянном тонусе. Поэтому пациенты с болезнью Томпсена имеют атлетический тип телосложения, однако мышечная сила снижена.
Дебют заболевания происходит достаточно рано, клинические проявления прогрессирует медленно, течение болезни имеет тенденцию к стабилизации.
- Миотония Беккера является более тяжелой формой врожденной миотонии по сравнению с болезнью Томпсена.
Патология манифестирует у детей в возрасте от 4 до 18 лет. Характерный мышечно-тонический феномен затрагивает не только дистальную мускулатуру, но вовлекает и проксимальные группы мышц конечностей. На поздней стадии поражаются мимические мышцы. Для пациентов характерны миалгии – мышечные болевые синдромы. В отличие от миотонии Томсена мышечные гипертрофии встречаются довольно редко.
Каналопатии с мутациями гена натриевого канала
Каналопатии с мутациями гена натриевого канала представлены калийзависимыми типами миотоний, врожденной парамиотонией и гиперкалиемическим периодическим параличом с миотонией.
- Калийзависимые миотонии включают несколько форм болезни, которые могут наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно.
По клиническим проявлениям заболевания являются схожими. Первые признаки возникают в возрасте от 5 до 55 лет. Основными симптомами болезни являются мышечные крампи, боли и скованность в мышцах, возникающие после напряжения или физической нагрузки. Наибольшая выраженность симптомов отмечается в нижних конечностях. Характерно возникновение гипертрофий мышц. Для калийзависимых миотоний также характерны длительные сокращения мышечных волокон в ответ на механическое воздействие извне. После подобного механического воздействия в период мышечного расслабления отмечается возникновение участков уплотнения в различных группах мышц. Наряду с этим часто выявляется патология сердечно-сосудистой системы – гипертрофическая кардиомиопатия и аритмии.
- Врожденная парамиотония (синоним болезнь Эленбурга, холодовая миотония) наследуется аутосомно-доминантно и характеризуется возникновением миотонического феномена под воздействием холода (при умывании холодной водой, прикладывание льда, употребление в пищу мороженного).
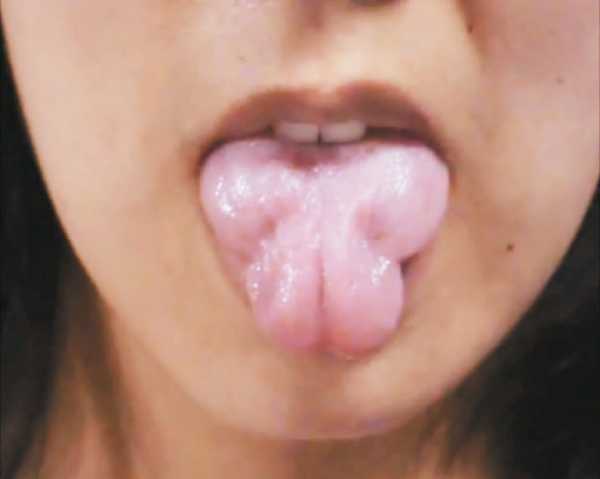
Наряду с этим имеет место внезапно возникающая мышечная слабость – транзиторный парез, длительность которого может достигать нескольких суток. Грубее всего страдает мимическая и жевательная мускулатура, мышцы языка, глотки, шеи. Кроме того, патологическим процессом могут быть затронуты и конечности. Повторные движения увеличивают выраженность мышечно-тонических проявлений. При согревании мышц миотонический синдром, напротив, уменьшается.
- Гиперкалиемический периодический паралич с миотонией характеризуется повторными эпизодами мышечной слабости, которая может быть как общей, так и локальной.
При этом подобные клинические проявления сочетаются с миотоническим феноменом. Наследование патологии аутосомно-доминантное. Дебют заболевания, как правило, приходится на первые 10 лет жизни. Эпизоды преходящей мышечной слабости могут быть спровоцированы употреблением пищи, богатой калием, чрезмерными физическими нагрузками, эмоционально-психическим стрессом, беременностью. Чаще всего миоплегия развивается в утренние часы. Слабость обычно начинается в дистальных отделах нижних конечностей и распространяется на туловище и верхние конечности. Продолжительность пароксизма мышечной слабости колеблется от нескольких минут до 1,5-2 часов. С возрастом частота и выраженность эпизодов миоплегии с миотонией имеет тенденцию к снижению.
Диагностика
Специфичность неврологических проявлений болезни обеспечивает возможность заподозрить миотонию, опираясь лишь на клинические симптомы. Однако золотым стандартом выявления миотонических синдромов является электронейромиография. При этом верификация диагноза требует проведения как игольчатой, так и стимуляционной электромиографии. Исследование позволяет зафиксировать патологическую возбудимость мембраны мышечных волокон в виде характерных мышечных разрядов высокой частоты.
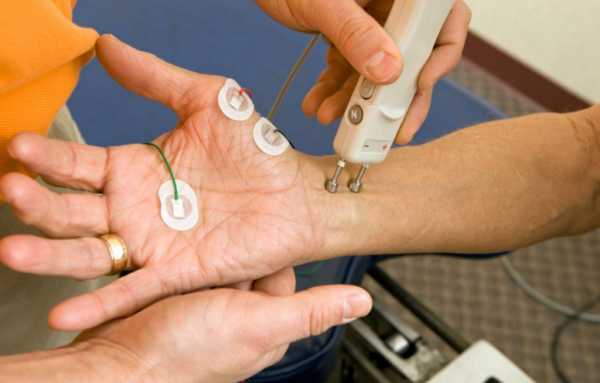
Точное установление формы болезни возможно только после проведения молекулярно-генетического анализа и выявлении мутантного гена.
Лечение
В настоящее время не существует эффективных методов воздействия на пораженные генные локусы. В этой связи единственным направлением в терапии пациента с миотонией является замедление прогрессирования болезни и достижение стойкой ремиссии. Для этого используют противосудорожные препараты, миорелаксанты, для снижения уровня калия – диуретики. Положительное влияние оказывают физиотерапевтические процедуры и лечебная физкультура. При сочетании миотонических феноменов с нарушением проведения нервного импульса могут быть использованы стимуляторы передачи нервного импульса (например, Нейромидин). Ранняя диагностика и адекватное лечение способны инициировать более мягкое, доброкачественное течение миотонических синдромов.
В современной медицине врожденные миотонические синдромы рассматривается как наследственная патология с отсутствием радикального эффективного лечения. Своевременная диагностика и грамотная терапия сохраняют трудоспособность и оптимальное качество жизни пациенту. Однако, учитывая генетическую природу патологии, лица, страдающие миотоническим синдромом, при планировании рождения ребенка должны в обязательном порядке пройти консультацию у врача-генетика.
Автор: Шоломова Елена Ильинична, неврологОцените эту статью:
Всего голосов: 89
4.27 89
Читайте также
Наследственные миотонические синдромы (миотония) - Неврология — LiveJournal
Наследственные миотонические синдромы (НМС) - группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами [1] дистрофических (ДМ) и [2] недистрофических миотоний (НДМ).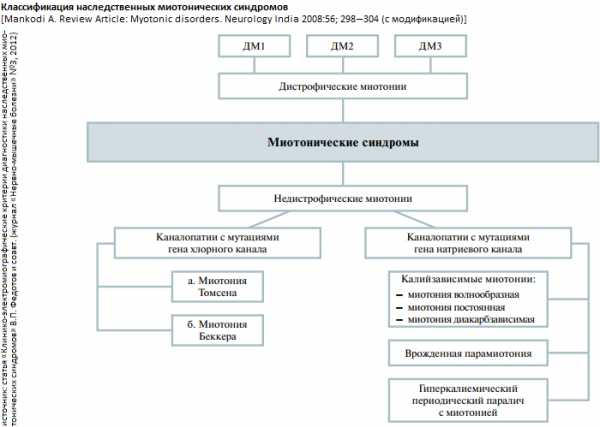
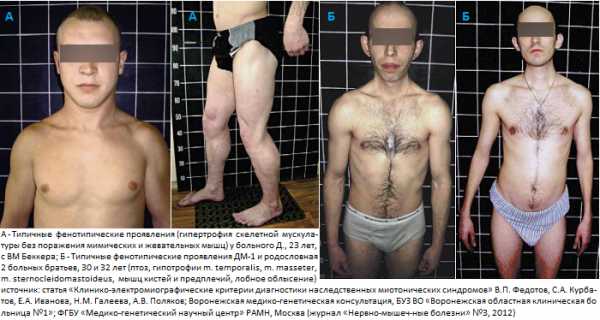
о клинической и ЭМГ феноменологии миотонии вы можете прочитать в статье «Клинико-диагностические критерии миотонии» Н. А. Шнайдер; ГБОУ ВПО Красноярский государственный медицинский университет имени проф. В. Ф. Войно-Ясенецкого Министерства здравоохранения РФ, кафедра медицинской генетики и клинической нейрофизиологии ИПО (журнал «Сибирское медицинское обозрение» №3, 2016) [читать]
Недистрофические миотонии. Самая распространенная форма НДМ - врожденная миотония (ВМ), с распространенностью от 1 до 9,4 на 100 тыс. населения, что зависит от страны и этнической принадлежности. Происхождение ВМ обусловлено мутациями в гене хлорного канала CLCN1 (локус 7q35) в поперечно-полосатых мышцах (нарушается проводимость ионов Cl- в клетку, что ведет к повышенной возбудимости мышечной мембраны и мышечной ригидности). ВМ клинически проявляются:
[1] генерализованными миотоническими феноменами;
[2] гипертрофией скелетной мускулатуры;
[3] транзиторной слабостью;
[4] дебютом в раннем возрасте;
[5] стационарным течением и благоприятным прогнозом.
На сегодня выделяют 2 формы ВМ. Первой описана миотония Томсена (МТ) с аутосомно-доминантным типом наследования и лишь 100 лет спустя - миотония Беккера (МБ) с аутосомно-рецессивным типом наследования. Молекулярно-генетические данные показали, что ранее считавшаяся редкой ВМ Беккера встречается даже чаще, чем ВМ Томсена. При дебюте в раннем возрасте пациенты с МТ и МБ имеют, как было указано выше, гипертрофию скелетных мышц («атлетическое сложение», более характерное для ВМ Томсена), иногда легкую миалгию, слабость, задержку мышечного расслабления, а также относительно стационарное течение и благоприятный прогноз. То есть больные с МТ и МБ имеют однотипные клинические проявления, различаясь лишь степенью их выраженности (ВМ Беккера считают несколько более тяжелой, но не в каждом случае).
Дистрофические миотонии (или ДМ, или сongenital myotonic dystrophy, myotonic dystrophy - DM) является мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Это наиболее распространенное заболевание из класса миотоний. Клиническая картина дистрофической миотонии складывается из 3 синдромов:
[1] миотонический синдром;
[2] дистрофический синдром;
[3] синдром вегетативно-трофических нарушений.
Таким образом, ключевая особенность ДМ - сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). МД характеризуется варьиру ющим началом заболевания: от пренатального периода до 50 - 60 лет. Различают четыре формы по возрастному «пику» начала заболевания: [1] врожденная (син.: конгенитальная ДМ или congenital myotonic dystrophy [CmyD]; клиническая симптоматика развивается сразу после рождения), [2] юношеская (син.: ювенильная МД или juvenile DM; с дебютом от 1 года до подросткового возраста), [3] классическая (син.: ДМ взрослых или adult DM; - с дебютом у индивидуумов старше 20, но моложе 40 лет) и [4] минимальная (син.: МД с поздним дебютом или DM with late onset; у индивидуумов старше 40 лет [как правило, до 60 лет] и с более легким течением). Это объясняется различиями в числе тринуклеотидных повто ров (CTG) в локусе гена МД, кодирующего синтез миотонин-протеинкиназы. Кроме различия в возрасте начала проявления болезни имеются достаточно различные клинические признаки разных подтипов этого заболевания.
До 1994 года ДМ считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: DM1 (мутация 19q13.3), DM2 (мутация 3q21) и DM3 (мутация 15q21-q24). Существуют отдельные исследования, подтверждающие наличие четвертого подтипа DM и др. (DM4, DMX). Наиболее часто встречается (среди всех миотоний, как среди ДМ, так и среди НДМ) DM1 - ДМ Россолимо-Куршмана-Штейнерта-Баттена (с частотой 13,5 на 100000 живых новорожденных; распространенность в больших популяциях около 1 : 8000). Распространенность DM2 (болезнь Thornton-Griggs-Moxley) и DM3 в настоящее время недостаточно изучена.
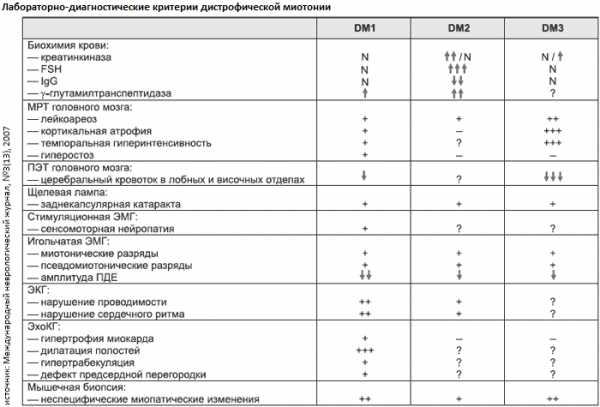
читайте также пост: Креатинкиназа (справочник невролога) (на laesus-de-liro.livejournal.com) [читать]
В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления ДМ вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с ДМ могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.
подробнее о DM1, DM2 и DM3 читайте [1] в статье «Клинико-генетическая гетерогенность дистрофической миотонии» Н.А. Шнайдер, Е.А. Козулина, Д.В. Дмитренко; Кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования ГОУ ВПО «Красноярская гос. мед. академия Федерального агентства по здраво-охранению и социальному развитию», Россия (Международный неврологический журнал, №3, 2007) [читать] (или [читать]) и [2] в статье «Миотоническая дистрофия. Современное представление и собственное наблюдение» Т.И. Стеценко, Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев, Украина (журнал «Современ-ная педиатрия» №1, 2014) [читать]
Обратите внимание! Для НДМ не характерно развитие парезов конечностей и мышечных атрофий в отличие от больных с ДМ1, у которых вялые дистальные парезы рук и ног служат главной причиной инвалидизации. Тем не менее, у больных с НДМ (форма Томсена, Беккера) специфическими симптомами могут выступать исходная неловкость и мышечная слабость в кистях, обусловленная типичными миотоническими задержками и исчезающая после нескольких повторных произвольных сокращений мышц. Симптом получил наименование «транзиторная слабость», а уменьшение выраженности миотонии при повторных мышечных сокращениях - «феномен врабатывания». Транзиторная слабость отсутствует у больных с ДМ1, а слабость в мышцах кисти остается неизменной даже после уменьшения миотонических проявлений. Наличие транзиторной слабости у больных с НДМ связывали с дебютом ДМ1, что затрудняло дифференциальную диагностику, приводило к неверным нозологическим трактовкам и неадекватным генетическим прогнозам в семьях пробандов. Все это послужило поводом к необходимости выработки объективных критериев наличия или отсутствия транзиторной слабости. Проведение теста с ритмической стимуляцией (РС) с частотой 10 - 60 Гц у больных с НМС выявило декремент М-ответа с восстановлением его амплитуды после тетанизации. Кроме того, при проведении РС у больных с НМС на примере мышц кисти, иннервируемых локтевым нервом, выявляется зависимость между величиной декремента амплитуды М-ответов и выраженностью транзиторной слабости. Выявленный декремент М-ответа при РС в генотипированной группе больных с НДМ объясняется нарушением функции хлорных каналов. [!!!] Таким образом, декремент амплитуды М-ответов может быть информативным показателем в алгоритме диагностического поиска мутаций в гене хлорного канала у пациентов с НМС.
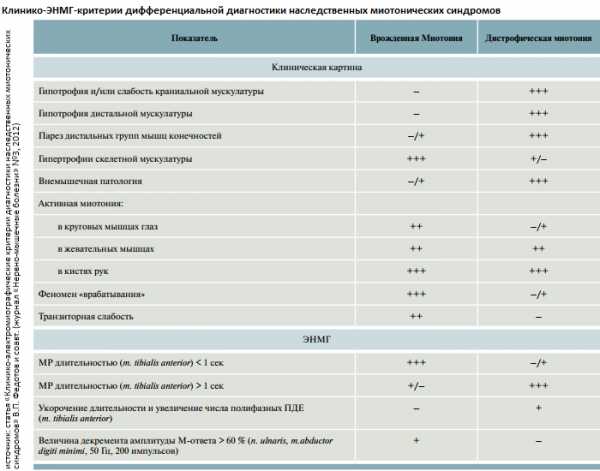
Читайте также:
статья «Миотоническая дистрофия: генетика и полиморфизм клинических проявлений» Е.О. Иванова, А.Н. Москаленко, Е.Ю. Федотова, С.А. Курбатов, С.Н. Иллариошкин; ФГБНУ «Научный центр неврологии», Москва, Россия; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр», Воронеж (журнал «Анналы клинической и экспериментальной неврологии» №1, 2019) [читать];
статья «Клинический случай дистрофической миотонии 1-го типа» Н.В. Ноздрюхина, А.А. Струценко, Е.Н. Кабаева, О. Тургунхужаев; РУДН, Москва (журнал «Трудный пациент» №3, 2019) [читать];
статья «Клинико-электромиографические критерии диагностики наследственных миотонических синдромов» В.П. Федотов, С.А. Курбатов, Е.А. Иванова, Н.М. Галеева, А.В. Поляков; Воронежская медико-генетическая консультация, БУЗ ВО «Воронежская областная клиническая больница №1»; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Нервно-мышеч-ные болезни» №3, 2012) [читать];
статья «Случай миотонии Беккера с псевдодоминантным типом наследования: современные подходы к дифференциальной диагностике миотоний Томсена и Беккера» С.А. Курбатов, С.C. Никитин, С.Н. Иллариошкин, П. Гундорова, А.В. Поляков; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; Региональная общественная организация Общество специалистов по нервно-мышечным болезням», Медицинский центр «Практическая неврология», Москва; ФГБНУ «Научный центр неврологии», Москва; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Нервно-мышечные болезни» №1, 2016) [читать];
статья «Случай миотонии Беккера с эквинной деформацией стоп» Г.Е. Руденская, Е.А. Иванова, Н.М. Галеева; Медико-генети-ческий научный центр РАМН, Москва (Журнал неврологии и психиатрии, №8, 2012) [читать];
статья «Миотонии» (сокращенное изложение) Teruyuki Kurihara, Internal Medicine 2005; 44:1027-1032 (подготовил Ю. Матвиенко) [читать];
диссертация на соискание ученой степени кандидата биологических наук «Генетическая вариабельность локуса миотонин-протеинкиназы в якутской популяции» Степанова С.К., ФГБНУ «Якутский научный центр комплексных медицинских проблем» ФГБНУ «Научно-исследовательский институт медицинской генетики»; Томск, 2015 [читать];
статья «Миотоническая дистрофия 2-го типа» Г.Е. Руденская, А.В. Поляков; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2012) [читать];
статья «Дистрофическая миотония Россолимо-Штейнерта-Куршмана» Авдей Г.М., Кулеш С.Д., Шумскас М.С., Авдей С.А.; УО «Гродненский государственный медицинский университет»; УЗ «Гродненская областная клиническая больница», г. Гродно (Уральский медицинский журнал, №9, 2014) [читать];
статья «Случай дистрофической миотонии 1-го типа с утяжелением клиники по линии отца» Курбатов С.А., Федотов В.П., Галеева Н.М., Забненкова В.В., Поляков А.В.; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; БУЗ ВО «Воронежская областная клиническая больница № 1»; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2015) [читать];
статья «Случай поздней диагностики дистрофической миотонии Россолимо-Штейнерта-Куршмана» Ю.Н. Быков, Ю.Н. Васильев, О.П. Панасюк, А.Ч. Янгутова; Иркутский государственный медицинский университет, кафедра нервных болезней (Сибирский медицинский журнал, №2, 2016) [читать];
статья «Клинико-генетическая гетерогенность хондродистрофической миотонии» Н.А. Шнайдер, Красноярский ГМУ им. проф. В.Ф. Войно-Ясенецкого, Красноярск (журнал «Нервно-мышечные болезни» №2, 2012) [читать];
статья «Профилактика аспирационной пневмонии у больных дистрофической миотонией с орофарингеальной дисфагией» Е.А. Бахтина, Н.А. Шнайдер, Т.Л. Камоза, Е.А. Козулина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого, кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования; Красноярский государственный торгово-экономический институт, кафедра технологии питания (журнал «Сибирское медицинское обозрение» №3, 2008) [читать];
статья «Мультидисциплинарный подход к ведению беременных, больных дистрофической миотонией» Н.А. Шнайдер, Е.А. Козулина, В.А. Шульман, С.Ю. Никулина, Р.А. Бутьянов, Е.А. Бахтина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого Росздрава; кафедра медицинской генетики и клинической нейрофизиологии ИПО; кафедра внутренних болезней №1 (журнал «Сибирское медицинское обозрение» № 4, 2008) [читать]